Fare hafızası, nöronlara özel dokuz harfli bir protein parçasına bağlanır için yorumlar kapalıGENETİK, sinirbilim
Hücrelerin bir ekleme adı verilen bir numarası vardır. Bir genin mesajını parçalara ayırabilir ve hangi parçaları saklayacaklarına karar verebilirler. Bu parçaları karıştırıp eşleştirerek, tek bir gen birçok farklı protein üretebilir, dokulara ve organlara gelişmek ve gelişmek için daha fazla seçenek sunar. Tüm dokular arasında, ekleme en çok beyinde yaygındır.
Çalışma, insan serebral korteksinde farklı nöron alt tiplerinin gelişimini yönlendiren gen ağlarını ortaya çıkardı Çalışma, insan serebral korteksinde farklı nöron alt tiplerinin gelişimini yönlendiren gen ağlarını ortaya çıkardı için yorumlar kapalıGENETİK, sinirbilim
İnsan beyninin, farklı rollere ve işlevlere sahip çok çeşitli hücre tipleri içerdiği bilinmektedir. Beyindeki hücrelerin, özellikle de en dış tabakasının (yani serebral korteks) yavaş yavaş uzmanlaştığı ve belirli roller üstlendiği süreçler, geçmiş birçok sinirbilim çalışmasının odak noktası olmuştur.
Sentetik Biyoloji: Yeni Yaşam Formları Yaratmanın Geleceği için yorumlar kapalıBİLİM, GENETİK, sağlık
Sentetik biyoloji, yaşamı anlama ve onunla etkileşim kurma biçimimizde dönüşüm yaratma potansiyeline sahip son teknoloji bir alan olarak ortaya çıkmıştır. Canlı sistemlerin yapısını ve işlevini anlamak, tasarlamak, yeniden yapılandırmak amacıyla mühendislik ilkelerini kullanır.Sentetik olarak yeni biyolojik bileşenlerin, devrelerin ve sistemlerin oluşturulduğu bir alandır.
Bu yazımızda, sentetik biyolojiyi çevreleyen heyecan verici olasılıkları ve etik hususları keşfedeceğiz.
Yaşamın yapı taşları: DNA sentezi ve genetik mühendisliği
Sentetik biyolojinin kalbinde DNA sentezi ve genetik mühendisliği yatar. Araştırmacılar, DNA dizilerini manipüle ederek yeni genetik kodlar tasarlayabilir ve oluşturabilirler. Bu sayede tamamen yeni organizmaların ortaya çıkmasına veya mevcut organizmaların geliştirilmesine olanak sağlanabilir. Bu süreç sayesinde bilim insanları, hastalığa karşı artan direnç veya gelişmiş üretkenlik gibi istenen özellikleri organizmalarda tanıtabilirler. Böylece, tıp ve tarımdan enerji ve çevre korumaya kadar uzanan alanlarda fırsatlar dünyasının kapısı aralanmış olur.
Mikroplardan mega yapılara: Sentetik biyolojinin uygulamaları
Sentetik biyoloji, birçok alanda ciddi bir potansiyele sahiptir. Tıpta, ilaç üretebilen veya belirli hastalıkları hedef alabilen genetiği değiştirilmiş mikroorganizmaların gelişmesine yol açabilir. Tarımda, sentetik biyoloji mahsul verimini optimize edebilir, besin içeriğini geliştirebilir ve sürdürülebilir tarım uygulamaları yaratabilir. Ayrıca sentetik biyoloji alanında, bilim insanlarının atıkları verimli bir şekilde biyoyakıtlara dönüştürebilen mikroorganizmalar tasarladıkları biyoenerji uygulamaları da vardır. Tüm bunlara ek olarak, sentetik biyoloji zararlı kimyasallara ve malzemelere biyo-temelli alternatifler yaratarak çevrenin korunmasında çok önemli bir rol oynayabilir.
Yaşam formları tasarlamak: Biyoinformatik ve bilgisayar modellemesi
Biyoinformatik ve bilgisayar modelleme, sentetik biyoloji alanının ayrılmaz bir parçasıdır. Bilim insanları, hesaplama araçlarının gücünden yararlanarak karmaşık biyolojik sistemleri simüle ve analiz ederler. Böylece, yeni yaşam formlarının fiziksel olarak yaratılmadan önce davranışları tahmin edilir. Sonuç olarak; tüm bu analizler, genetik tasarımların optimizasyonuna izin verir, deneme yanılma deneylerinde zaman ve maliyeti azaltır. Bilgisayar biliminin sentetik biyoloji ile entegrasyonu, canlı organizmalar üzerinde daha verimli ve hassas mühendisliğin yolunu açar.
Etik hususlar ve önümüzdeki zorluklar
Sentetik biyolojinin potansiyeli yüksek olsa da önemli etik kaygıları da beraberinde getirir. Bilim insanları yeni yaşam formları yaratma becerisi kazandıkça güvenlik, ekolojik etki ve teknolojinin sorumlu kullanımı ile ilgili sorular ön plana çıkar. Yenilik ve tedbir arasında bir denge kurmak çok önemlidir. Sentetik biyolojinin etik ve sorumlu bir şekilde geliştirilmesini sağlamak için; bilim insanları, politika yapıcılar ve halk arasında sağlam düzenlemeler, risk değerlendirmeleri ve açık diyalog gereklidir.
Sentetik biyoloji, yaşamı tasarlama yeteneğimizde ileriye doğru dönüştürücü bir sıçramayı temsil eder. Araştırmacılar, farklı disiplinlerden ilkeleri birleştirerek arzu edilen özelliklere sahip yeni organizmalar yaratmanın mümkün olan sınırlarını zorlarlar. Ancak, gelişmekte olan bu alanda yol alırken, içerik oluşturucu rolünü oynamanın getirdiği etik kaygıları ve zorlukları ele almak çok önemlidir. Sorumlu düzenleme ve açık diyalog yoluyla, insanlık ve gezegen için daha iyi bir geleceği şekillendirirken, sentetik biyolojinin gücünden yararlanabiliriz.
Nadir görülen genetik bozukluk ilk kez rahimde tedavi edildi için yorumlar kapalıGENETİK
Şu anda neredeyse üç yaşında olan çocukta, sıklıkla ölümcül olan motor nöron hastalığının hiçbir belirtisi görülmüyor.
İki buçuk yaşındaki bir kız, rahimdeyken motor nöron rahatsızlığı için tedavi gören ilk kişi olduktan sonra nadir görülen bir genetik bozukluğun hiçbir belirtisini göstermiyor. Çocuğun annesi, hamileliğin sonlarında gen hedefli ilacı aldı ve çocuk da almaya devam ediyor.
Avustralya’daki UNSW Sydney’de pediatrik nörolog olan Michelle Farrar, “Bebek etkili bir şekilde tedavi edildi ve hastalığın hiçbir belirtisi görülmedi” diyor. Sonuçlar dün New England Journal of Medicine’de yayımlandı.
Çocuk, hareketi kontrol eden motor nöronları etkileyen ve ilerleyici kas zayıflamasına yol açan spinal musküler atrofi olarak bilinen genetik bir rahatsızlıkla gebe kaldı. Yaklaşık her 10.000 doğumdan birinde bu rahatsızlığın bir türü görülür — bu da onu bebeklerde ve çocuklarda önde gelen genetik ölüm nedeni yapar.
En şiddetli formunda, bu çocukta olduğu gibi, bireyler SMN1 geninin her iki kopyasından da yoksundur ve bu eksikliği kısmen telafi eden komşu bir gen olan SMN2’nin yalnızca bir veya iki kopyasına sahiptir . Sonuç olarak, vücut omurilik ve beyin sapındaki motor nöronlarını korumak için gereken proteini yeterince üretmez. Bu protein, ikinci ve üçüncü trimesterlerde ve yaşamın ilk birkaç ayında en önemlidir. Şiddetli hastalığı olan bebekler genellikle üçüncü yaş günlerinden sonra yaşamazlar.
Geçtiğimiz on yılda, ABD Gıda ve İlaç Dairesi (FDA) spinal musküler atrofi için yenidoğanları tedavi etmek üzere üç ilacı onayladı. Bu çalışmada kullanılan Risdiplam adlı oral ilaç, İsviçre’nin Basel kentinde bulunan biyoteknoloji firması Roche tarafından üretilen, SMN2 geninin ifadesini değiştirerek daha fazla SMN proteini üreten küçük bir moleküldür.
Şimdiye kadar spinal musküler atrofi için tedaviler doğumdan sonra uygulanıyordu. Ancak SMN1 geninin her iki kopyasından da yoksun ve SMN2 geninin yalnızca iki kopyasına sahip yenidoğanların yarısına kadarı bazı semptomlarla doğuyor. Çalışmaya liderlik eden, Tennessee, Memphis’teki St. Jude Çocuk Araştırma Hastanesi’nde klinik sinir bilimci olan Richard Finkel, “Hala iyileştirme için yer vardı” diyor.
Ebeveyn önerisi
Finkel, ilacın rahimde verilmesi fikrinin ebeveynlerden geldiğini söylüyor. “Bu korkunç hastalıktan dolayı zaten bir kayıp yaşamışlardı,” diyor ve doğumdan önce başlayabilecekleri tedavi seçenekleri olup olmadığını bilmek istiyorlardı. FDA bu birey için çalışmayı onayladı.
32 haftalık hamile olan anne, altı hafta boyunca her gün Risdiplam aldı. Bebek ilacı yaklaşık bir haftalıkken almaya başladı ve muhtemelen hayatının geri kalanında almaya devam edecek.
İnsanlar orta yaş itibarıyla tamamen değil ama çoğunlukla tümüyle birbirinin aynı olmayan karmaşık birer hücre mozaiğidir. Görsel: Knowable Magazine
Vücudunuzu gözünüzün önüne getirin: Vücudunuz ömür boyunca biriken binlerce genetik hata taşıyan bir hücre koleksiyonudur. Bu hataların pek çoğu zararsız, bazıları zararlıdır ve en az birkaçı da sizin için iyi olabilir.
Yumurta ve sperm birbiriyle buluştuğunda ve biyolojik ebeveynlerinizin DNA’sı beraber bir takım oluşturduğunda başladınız. İlk hücreniz yeni harmanlanmış genomunu kopyalamaya ve bölünerek bir vücut inşa etmeye başladı.
Ayrıca neredeyse hemen genetik hatalar birikmeye başladı.
İngiltere Hinxton’daki Wellcome Sanger Enstitüsünde kanser biyoloğu olarak çalışan Phil H. Jones, “Bu hataların genomunuzda birikme süreci yaşam boyu devam eder” diyor.
Bilim insanları DNA kopyalama sistemlerinin ara sıra hata yaptığını uzun süredir bilse de (kanserler sık sık böyle başlıyor); teknoloji sadece kısa süre önce her genetik yanlışın sınıflandırılacağı kadar hassas hale geldi. Üstelik hatalarla dolu olduğumuz ortaya çıktı. Her insan, bir hücre veya hücre grubundan diğerine kadar çoğunlukla birbirinin aynısı olup tek tük farklılıklar gösteren devasa bir hücre mozaiği.
Hücresel genomlar bir noktada tek bir genetik harf ile ve başka bir noktada ise kayıp olan daha büyük bir kromozom parçasıyla farklılık gösterebilir. Worcester’daki Massachusetts Chan Tıp Fakültesinde çalışan moleküler biyolog Michael Lodato, orta yaş itibarıyla vücudun her hücresinin muhtemelen bin kadar genetik yazım hatası barındırdığını tahmin ediyor.
Bu mutasyonlar (ister kanda olsun, ister ciltte veya beyinde), hücrenin DNA kopyalama mekanizması olağanüstü derecede isabetli olsa ve hücreler mükemmel onarım mekanizmaları sergilese bile ortaya çıkıyor. Yetiştkin bir vücut, 4 milyonu her saniye bölünen yaklaşık 30 trilyon hücre içerdiğinden, nadir hatalar bile zamanla birikiyor. (Hatalar, yumurta ve spermleri ortaya çıkaran hücrelerde çok daha seyrek; görünüşe göre vücut, mutasyonları üreme dokularından uzak tutmak için daha çok çaba ve enerji harcıyor ki gelecekteki nesillere bozulmamış DNA aktarılsın.)
“Hepimizin bu kadar iyi devam ediyor olması küçük bir mucize” diyor Jones.
Mutasyon barındıran hücrelerin bir dokuyu ele geçirmesi sonucunda hastalık ortaya çıkacak diye bir kural yok. Klonların genişlemesini teşvik eden mutasyonlar, kansere yön veren tehlikeli etmenler olabilir fakat bunlar nötr de olabilir. Hatta bir dokunun bütünlüğünü sürdüren ve kanseri teşvik etmeyen faydalı mutasyonlar bile olabilir. Görüntü: Knowable Magazine
Bilim insanları bu mutasyonların sebep ve sonuçlarını araştırmanın hâlâ erken aşamalarında. ABD Ulusal Sağlık Enstitüleri, bunların kataloglanması için 140 milyon dolar sermaye ayırmış. ABD Ulusal Zihin Sağlığı Enstitüsü ise beyindeki mutasyonlarınincelenmesine on milyonlarca dolar harcıyor. Pek çok değişim muhtemelen zararsız olsa da bazıları, kanserler ve nörolojik hastalıklar bakımından sonuçlar doğurabilir. Daha temelinde ise bazı araştırmacılar, yaşlanma sürecinin büyük bir kısmının altında ömür boyu gerçekleşen rastgele genomik hataların olabileceğinden şüpheleniyor.
“Bunu bileli on yıl bile olmuyor ve yeni bir kıtanın keşfedilmesine benziyor” diyor Jones. “Bütün bunların ne anlama geldiği konusunda henüz yüzeysel şeyler biliyoruz.”
Baştan şüpheli
Bilim insanları üreme dışı veya somatik dokularda biriken genetik yazım hataları ve diğer mutasyonların, hastalıkları ve yaşlanmayı açıklamaya yardımcı olabileceğinden DNA’nın yapısının keşfedildiği 1950’lerden bu yana şüpheleniyor.
1970’lerde araştırmacılar, hücrelerin bir bölümünde büyümeyi teşvik eden mutasyonların kanserlerin kökeni olduğunu biliyordu.
New York’taki Albert Einstein Tıp Fakültesinde çalışan genetikçi Jan Vijg, “Bu olayın frekansının çok ama çok düşük olduğu varsayılıyordu” diyor.
Fakat bu mutasyonları tespit edip incelemek son derece zordu. Standart DNA dizilemesinde, sadece geniş hücre gruplarından çıkarılan büyük miktarlardaki genetik malzeme analiz edilebiliyor ve sadece en yaygın dizilimler ortaya çıkarılıyordu. Nadir mutasyonlar ise radara girmiyordu. Kaliforniya’daki Stanford Üniversitesinde çalışan kök hücre biyoloğu Siddharta Jaisval, bu durumun 2008 civarında değişmeye başladığını söylüyor. Yeni teknikler o kadar hassas ki, hücrelerin ufak bir kısmında (hatta tek bir hücrede) bulunan mutasyonlar bile ortaya çıkarılabiliyor.
Jaisval 2010’ların başlarında, mutasyonların kan kanserine dönüşmeden önce insanların kan hücrelerinde nasıl birikebildiğiyle ilgileniyormuşb. 17.000’den fazla kişinin kanını inceleyen Jaisval ve meslektaşları, tahmin ettikleri şeyi bulmuş: Kanserle ilişkili mutasyonlar 40 yaşın altındakilerde nadirken, yaş ile beraber daha yüksek miktarlarda görülüyor ve 70’nci yaş gününden sonra kan hücrelerinin yaklaşık yüzde 10’u veya daha fazlasını oluşturuyorlarmış.
Fakat araştırma takımı, genelde mutasyonlu hücrelerin genetik olarak birbirinin aynısı olduğunu da görmüş: Bunlar klonmuş. Jaisval bunun sebebinin, vücudun kan üreten binlerce kök hücresinden birinin, büyüme ve bölünmede kendini biraz daha iyi hale getiren mutasyonları seçmesi olduğunu düşünüyor. Bu hücre normal şekilde büyüyen kök hücrelere onlarca yıl içerisinde galip gelerek, genetik olarak uyumlu büyük bir hücre grubu meydana getiriyor.
Etkili biçimde bölünen bu mutasyonlu kan hücresi klonları, doğal olarak kan kanseri tehlikesiyle ilişkilendirilmiş. Fakat aynı zamanda kalp hastalığı, inme ve herhangi bir sebepli ölümde risk artışıyla da (belki de enflamasyonu teşvik ettikleri için) ilişkilendirilmişler. Ayrıca beklenmedik bir şekilde, Alzheimer demansı riskinin üçte bir kadar azalmasıylailişkilendirilmişler. Kan hücresi klonlarının sağlığa etkileri üzerine 2023 Annual Review of Medicine bülteninde yayımlanan bir makalenin eş yazarlığını yapan Jaisval, bazı klonların beyin dokusunda çoğalma veya toksik proteinleri temizleme konusunda daha iyi olabileceğini düşünüyor.
Jaisval ve meslektaşları 2014 yılında rapor ettikleri bu kan klonlarını takip ederken, Wellcome Sanger Enstitüsünde çalışan araştırmacılar da göz kapağı derisiyle başlayarak diğer dokulardaki vücut mutasyonlarını incelemeye başlamışlar. Bazı insanların göz kapakları yaş ile birlikte sarkıyor ve bu sorunun düzeltilmesi için bir miktar cilt ameliyatla alınıyor. Araştırmacılar dört bireyden bu parçaları alarak, genetik dizileme için 1 ya da 2 milimetrelik daireler kesmiş. Wellcome Sanger’da çalışan genetikçi Inigo Martincorena, “Sürprizlerle doluydu” diyor. Hastalarda cilt kanseri olmasa da ciltleri binlerce klonla doluymuş ve göz kapağı cilt hücrelerinin beşte birinden üçte birine kadarı, kanserle ilişkili mutasyonlar barındırıyormuş.
Cilt kanseri olmayan insanlardaki bu kadar fazla cilt kanserinde mutasyon olduğunu gösteren bulgular büyük ilgi çekmiş. Colorado Üniversitesi Tıp Fakültesinde kanser biyoloğu olarak çalışan ve söz konusu çalışmada yer almayan James DeGregori, “Ağzım acık kalmıştı” diyor.
Wellcome Sanger araştırmacıları ise yemek borusu, mesane ve kalın bağırsağın da içinde bulunduğu diğer çeşitli dokularda birbirinin aynısı olan, mutasyon geçirmiş hücreleri belirlemeye başlamışlardı. Örneğin, bağırsak duvarındaki girintiler olan kolonik girintileri incelemişlerdi. Bunlardan her insanda yaklaşık 10 milyon tane var ve her biri yaklaşık 2.000 hücre barındırıyor. Bu hücrelerin de her biri, o girintide yer alan bir avuç kök hücresinden ortaya çıkıyor. 42 kişide 2.000’den fazla girintinin incelendiği bir çalışmada araştırmacılar, 50’li yaşlarda olan insanların girinti yapılarında yüzlerce genetik varyasyon bulmuş.
Bu yaş grubunda diğer türlü normal olan girintili yapıların yüzde 1 kadarı, kanserle bağlantılı mutasyonlar içeriyormuş. Söz konusu mutasyonların bazıları, civardaki hücrelerin yayılmasını baskılayarak mutant hücrelerin girinti yapısını daha çabuk ele geçirmesineimkan tanıyor. Bu durum tek başına kalın bağırsak kanseri oluşturmak için yeterli olmayabilir ama nadir vakalarda hücreler, kansere sebep olan ek mutasyonlar kazanarak girinti yapısının sınırlarından taşıp kötü huylu tümörlere sebep oluyor.
“İnsanlar bu somatik mutasyonları her yerde aradı ve onları her organda bulduk” diyen Jones, vücudu bir çeşit evrimsel savaş meydanı gibi görüyor. Hücreler mutasyon biriktirdikçe, daha fazla (veya daha seyrek) büyüyüp bölünebiliyorlar. Daha kolay çoğalan bazı hücreler, zamanla diğerlerini geride bırakabiliyor ve geniş klonlar oluşturabiliyor.
“Ayrıca” diye belirtiyor DeGregori, “yumrulu bir şeye dönüşmüyoruz.” Dokularımızın, klonların kansere dönüşmesini engelleyen yöntemleri olması gerektiğini ileri sürüyor. Aslında Jones ve diğer makale yazarının 2023 Annual Review of Cancer Biology bülteninde tarif ettikleri üzere, farelerde fazla büyüyen mutant klonların normal büyümeye geri döndüğü görülmüş.
Jones ve meslektaşları, insanlardaki yemek borusunda bir koruma örneği keşfetmişler. Genelde yemek borusu dokusunun büyük bir kısmını oluşturan pek çok klon, orta yaş itibarıyla NOTCH1 adı verilen bir geni bozan mutasyonlara sahip. Bu durum yemek borusunun yiyecekleri taşıma kabiliyetini etkilemiyor ama kanserlerin büyümek için NOTCH1‘e ihtiyacı olduğu görünüyor. Yemek borusu hücrelerinde kötü mutasyonlar birikebiliyor fakat NOTCH1 olmadığında, tümöre dönüşme ihtimalleri daha düşük gibi duruyor.
Diğer bir ifadeyle bedensel mutasyonların bazıları kötü veya nötr değil, hatta faydalı. Ayrıca neyse ki bu iyi mutasyonlar çoğu zaman galip geliyor.
Beynin içerisine girmek
DNA koplama mekanizmamızın yemek borusu, kalın bağırsak ve kan hücrelerinde hata yapma bakımından pek çok olanağı var çünkü bunlar sürekli bölünüyor. Fakat beynimizdeki nöronlar, doğumdan önce veya kısa süre sonra bölünmeyi bırakıyor. Boston Çocuk Hastanesinde nörogenetikçi olarak çalışan Christopher Walsh, bu yüzden bilim insanlarının esasında bunların genetik açıdan bozulmamış kaldıklarını varsaydıklarını söylüyor.
Fakat yaşam boyunca biriken mutasyonların beyinde problemlere sebep olabileceğini gösteren işaretler var. Araştırmacılar 2004 yılında, sadece bazı beyin hücrelerinde bulunan bir mutasyon sebebiyle Alzheimer hastalığı olan bir hasta rapor etmiş. Bu mutasyon yeniymiş ve her iki ebeveynden de geçmemiş.
Ayrıca 2012 yılında Walsh’ın grubu, beyinde fazla büyüyen ve nöbetlere sebep olan bir yeri düzeltmek için yaptıkları ameliyat sırasında alınan beyin dokusu üzerinde bir analiz yürütmüş. Sekiz numuneden üçünde, beyin boyutunu düzenleyen bir geni etkileyenmutasyonlar varmış ancak bu mutasyonların kanda devamlı bulunmaması, vücudun sadece bir kısmında ortaya çıktıklarını akla getiriyor.
Lodato, beyin hücrelerinin çeşitli şekillerde mutasyon kazanabileceğini söylüyor. Bir mutasyon gelişimin erken aşamasında, beyin tamamlanmadan ve hücreleri bölünmeyi durdurmadan önce ortaya çıkabilir. Ya da erişkin bir beyin hücresinde DNA hasar görebilir ve düzgün onarılmayabilir.
2012 yılında kalıtımla geçmeyen beyin mutasyonlarına olan ilgi artmaya başlamış. O zamanlar ABD Ulusal Zihin Sağlığı Enstitüsü başkanı olan Thomas Insel, pek çok psikiyatrik durumun altında bu tür mutasyonların yatıyor olabileceğini öne sürmüş. Beyindeki bu katılımla alınmamış mutasyonlar, nörolojik hastalıklarda uzun süredir var devam eden bir bilmeceyi açıklığa kavuşturabilir: Mesela neden tek yumurta ikizlerine genelde aynı psikiyatrik teşhisler konmuyor? (örneğin ikizlerden birinde şizofreni gelişirse, diğerinde hastalığın ortaya çıkma ihtimali sadece yüzde 50 kadar).
San Diego’da bulunan ve epilepsinin şiddetli bir türüyle ilgili yapılan araştırmaları ve aileleri destekleyen, kâr amacı gütmeyen Lennox-Gastaut Sendromu Vakfı’nın bilimsel direktörü sinirbilimci Mike McConnell, mozaikliğin “çok ikna edici bir cevap sunduğunu” söylüyor.
McConnell, Walsh, Lodato ve diğerleri, 2010’ların başlarından itibaren hayatını kaybeden insanların beyinlerine serpilmiş büyüklü küçüklü mutasyonları sınıflandırmaya başlamışlar. Genlerin, birden fazla genin veya kromozomların tamamının delesyon ve kopyalarının tek tek çetelesini tutmuş; genomda yeni noktalara giden kromozom bölümlerini bütünüyle belirlemişler. Walsh, Lodato ve meslektaşları nihayetinde, 50’li yaşlardaki kişilerin her bir sinir hücresinin genetik kodunda bin veya daha fazla tek harifli mutasyon olduğunu keşfetmiş. O son bulgunun “Tamamen imkansız göründüğünü” anımsıyor Walsh. “Kendimizden şüphe duymuştuk.”
Böylesine çarpıcı sonuçların karşısında bilim insanları daha da fazlasını araştırmış. Yaşları dört aylık ile 82 arasında değişen ve vefat etmiş 15 kişiden alınan 159 nörona bakmışlar. Araştırmacılar mutasyon sayılarının yaşla beraber arttığını ve bu durumun da hataların, vücudun diğer kısımlarında olduğu gibi zamanla arttığını gösterdiğini bildiriyor. “Beyin, büyük ve derin biçimde bir mozaik” diyor Lodato.
ABD Ulusal Zihin Sağlığı Enstitüsü, bu mozaikliği daha da fazla araştırmak için 2015’ten 2019’a kadar yürütülen bir dizi projeye fon sağlamış. Bu projelerde, genellikle ölümden sonra toplanan ve doku bankalarına yatırılan, nörotipik veya Tourette sendromu ve otizm tayf bozukluğu gibi durumlara sahip 1.000’i aşkın kişiye ait numunelerde beyin dokusunun mozaikliği araştırılmış.
Projenin eş öncülüğünü yürüten McConnell, tek harfli mutasyonların en yaygın olduğunu söylüyor. Araştırmacılar 400 terabaytı aşkın DNA dizilim verisi ve başka veriler toplayıpanalitik araçlar geliştirerek, beyin mozaikliği çalışmalarında bir sonraki turun inşa edileceği güçlü bir platform oluşturmuş. Bilim insanları bu ve diğer çalışmalardan, beyin mozaikliği ile otizm, epilepsi ve şizofreni gibi nörolojik hastalıkları ilişkilendirmiş.
Lodato’nun laboratuvarında çalışan yüksek lisans öğrencileri Cesar Bautista Sotelo ve Sushmita Nayak, biriken mutasyonların felç edici bir durum olan amiyotrofik lateral skleroza nasıl sebep olabileceğini araştırıyor. Genetikçiler, kalıtımsal olmayan vakaların sadece yüzde 10 kadarında bilinen bir mutasyon belirleyebiliyor. Fakat mozaikliğe yönelik yeni veriler, daha pek çok insanın beyinleri veya omuriliklerindeki ALS genlerinde mutasyonlar olabileceğini akla getiriyor; bunlar vücutlarının geri kalanında olmasa bile.
Bu önemli çünkü bilim insanları, mutasyon geçirdikleri zaman ALS’ye sebep olan 40’ı aşkın genin bazılarının hedef alındığı terapiler üzerinde çalışıyor. Gıda ve İlaç Dairesi, mutasyon geçiren yaygın bir ALS genini kapatan böyle bir tedaviyi 2023 yılında ilk defa onaylamıştı. Hastaların bu gibi terapilere hak kazanması için mutasyonlarını bilmeleri gerekecek.
Bu yüzden, diyor Nayak, “ALS teşhisindeki mevcut uygulamanın değiştirilmesini güçlü biçimde savunuyoruz.” Sadece bir kan numunesindeki DNA’ya bakmak yerine, kanı değil de vücuttaki dokuları meydana getiren hücrelerin gelişimi esnasında bir ALS mutasyonu ortaya çıkarsa diye tükürük, saç veya cilt gibi diğer dokular da incelenebilir.
Nasıl yaşlandığımıza dair ipuçları
Şimdilik, vücudumuzdaki mozaikliğin sağlık açısından doğurduğu sonuçlar çoğunlukla eylem gerektirecek kadar net değil; özellikle de sunulacak alakalı bir tedavinin olmadığı kan klonları gibi durumlarda. “İnsanların bu konuda endişelenmesi gerektiğini savunmuyoruz aslında” diyor Jaiswal. “Zamanın bu noktasında, sağlıklı olan insanları test etmenin bir mantığı yok.”
Martincorena ve meslektaşları, 2022 tarihli bir çalışmada bu kuramın öğelerinden birini test etmiş. Eğer mutasyon birikimi yaşlanmaya katkıda bulunuyorsa, o halde fare gibi kısa ömürlü mutasyonlar hızlı birikirken insanlar gibi daha uzun ömürlü türlerde, belki de onarım mekanizmalarının daha iyi olması sebebiyle mutasyonların daha yavaş biriktiğini öne sürmüşler.
Araştırmacılar bu fikri araştırmak üzere, sekiz insan ve ayrıca bir canlı koleksiyonundan alınan kalın bağırsak girinti numunelerini inceledikleri beş yıllık bir yolculuğa çıkmışlar. Söz konusu canlılar arasında 19 laboratuvar faresi ve sıçan; kediler, köpekler, inekler ve tavşanlar gibi 15 evcil hayvan; kaplanlar, lemurlar, bir liman yunusu ve 30 yıldan fazla yaşamasıyla bilinen tüysüz köstebek farelerinin yer aldığı 14 egzotik hayvan daha var. Tahmin edildiği gibi tür ne kadar uzun yaşıyorsa, mutasyonların birikimi de o kadar yavaşoluyor.
Araştırmacılar birkaç türe ait kalın bağırsak girintisini analiz ederek, daha uzun yaşayan türlerde mutasyonların daha yavaş biriktiğini belirlemiş. Bu durum, yaşlanmayla ilişkilendirilen vücut hücresi mutasyonlarına dair uzun süredir var olan bir kuram ile tutarlılık gösteriyor.
“Bu durum somatik mutasyonların yaşlanmaya sebep olduğunu göstermiyor fakat en azından bir miktar rol oynadıkları ihtimaliyle tutarlılık sergiliyor” diyor Martincorena. Bu noktada iş başında olan iki unsur var: Mutasyonların birikmesi ömrün daha kısa olmasına katkıda bulunuyor fakat sonrasında bu kısalan ömür, mutasyon korumasının önemini azaltıyor; dolayısıyla kısa ömürlü türlerde, DNA onarımına daha düşük yatırım oluyor.
Mutasyonların yaşlanmaya katkıda bulunabileceği fikri, onları mağlup etmenin genetik bir gençlik çeşmesi olabileceğini akla getiriyor. “Eğer yarın öbür gün bu mutasyonların birikmesini durdurmanın bir yolunu bulursam, inanılmaz zengin olurum” diyor Bautista Sotelo. Halihazırda en az bir biyoteknoloji startup’ı (New York’taki Matter Bio), insan genomunu onarma hedefiyle fon toplamış. (Böyle bir planın o kadar geniş bir hücre yığınında uygulanıp uygulanamayacağı başka bir mesele: “Mutasyonlardan kurtulabileceğinizi sanmıyorum” diyor DeGregori.)
Vücuttaki mutasyonların hikayesi sona ermekten çok uzakta. “Şu an yaptığımız keşiflere bakarsak, yolculuk daha yeni başladı” diyor Martincorena. “Önümüzdeki birkaç yıl içinde pek çok sürpriz bekliyorum.”
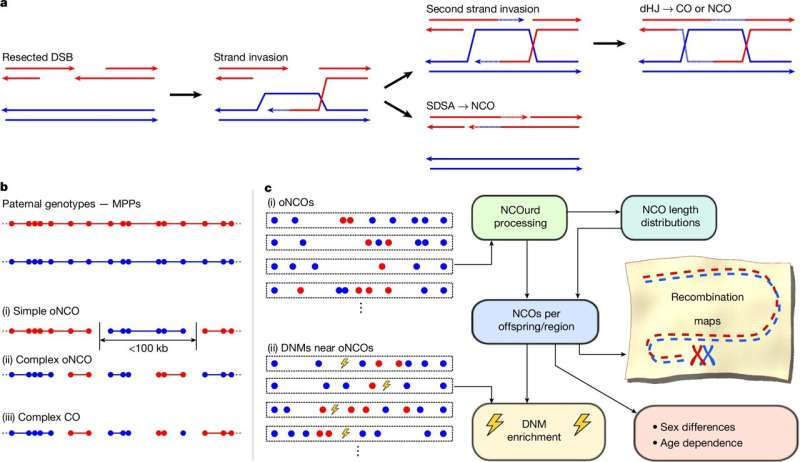
İnsan genomunun tam rekombinasyon haritası oluşturuldu için yorumlar kapalıGENETİK, Rekombinant
deCODE genetics/Amgen’deki bilim insanları, insan DNA’sının üreme sırasında aktarılırken nasıl karıştırıldığına dair eksiksiz bir harita oluşturdular. Harita, genetik çeşitliliğin ve sağlık ve doğurganlık üzerindeki etkisinin anlaşılmasında önemli bir adım teşkil ediyor. deCODE genetics’te insan genomunda yeni çeşitliliğin nasıl oluşturulduğu ve sağlık ve hastalıkla ilişkisi üzerine 25 yıllık araştırmanın devamı niteliğinde.
Bugün Nature dergisinin çevrimiçi baskısında yer alan yeni harita , yüksek DNA dizi benzerliği nedeniyle tespit edilmesi zor olan büyük ebeveyn DNA’sının daha kısa ölçekli karıştırılmasını (çaprazlama olmaması) içeren ilk haritadır. Harita ayrıca, kritik genetik işlevleri korumak veya kromozomal sorunları önlemek için büyük olasılıkla büyük yeniden karıştırmadan yoksun olan DNA alanlarını da belirler. Bu içgörü, bazı gebeliklerin neden başarısız olduğuna ve genomun çeşitliliği istikrarla nasıl dengelediğine dair daha net bir resim sunar.
Rekombinasyon olarak bilinen bu karıştırma , genetik çeşitlilik için elzem olsa da, süreçteki hatalar ciddi üreme sorunlarına yol açabilir. Bu başarısızlıklar, gebeliklerin devam etmesini engelleyen genetik hatalara yol açabilir ve kısırlığın dünya çapında yaklaşık on çiftten birini etkilemesinin nedenini açıklamaya yardımcı olabilir. Bu süreci anlamak, doğurganlık tedavilerini iyileştirmek ve gebelik komplikasyonlarını teşhis etmek için yeni umutlar sunar.
Araştırma ayrıca erkekler ve kadınlar arasında genom rekombinasyonunun nasıl ve nerede gerçekleştiği konusunda önemli farkları ortaya koyuyor. Kadınlarda daha az çapraz olmayan rekombinasyon var ancak bunların sıklığı yaşla birlikte artıyor ve bu da daha yaşlı anne yaşının neden daha yüksek gebelik komplikasyonları ve çocukta kromozomal bozukluk riskleriyle ilişkili olduğunu açıklamaya yardımcı olabilir. Ancak erkekler bu yaşa bağlı değişimi göstermiyor ancak her iki cinsiyetteki rekombinasyon yavrulara geçen mutasyonlara katkıda bulunabilir.
Rekombinasyon sürecini anlamak, insanların bir tür olarak nasıl evrimleştiğini ve sağlık sonuçları da dahil olmak üzere bireysel farklılıkları neyin şekillendirdiğini anlamak açısından da önemlidir. Tüm insan genetik çeşitliliği, rekombinasyona ve de novo mutasyonlara, yani çocukta bulunan ancak ebeveynlerde bulunmayan DNA dizilerine kadar izlenebilir. Harita, mutasyonların DNA karışımının olduğu bölgelere yakın yerlerde arttığını ve dolayısıyla bu iki sürecin oldukça ilişkili olduğunu göstermektedir.
Uzun okuma dizilemesi, nadir hastalık teşhislerinin zamanını ve maliyetini azaltırken daha fazla genetik bilgi ortaya çıkarıyor için yorumlar kapalıGENETİK, hastalıklar, Sendromlar
Dünya çapında her 10 kişiden biri nadir görülen bir genetik hastalıktan etkileniyor ancak genetik teknoloji ve testlerdeki hızlı artışlara rağmen bunların yaklaşık %50’si teşhis edilemiyor. Bir kişi teste erişebilse bile, teşhis alma süreci yaklaşık beş yıl veya daha fazla sürebilir ve bu da çoğu zaman çocuk olan hastalar için doğru tedaviye başlamak için bazen çok geç olabilir.
Bunun bir nedeni de, mevcut klinik testlerin, genomun belirli bölgelerindeki bilgilere erişemeyen ve bu nedenle tanı koymaya yardımcı olabilecek önemli kanıtları gözden kaçırabilen kısa okuma dizilemesi adı verilen bir yöntemi kullanmasıdır.
Ancak UC Santa Cruz araştırmacıları, varyasyonu bulmak için daha kapsamlı bir veri seti sağlayabilen, birden fazla özel teste olan ihtiyacı ortadan kaldırabilen ve nadir hastalıkların teşhisini kolaylaştırabilen uzun okuma dizilemesi adı verilen son teknoloji ürünü alternatif bir yöntem üzerinde araştırmaları ilerletiyorlar.
Yeni bir araştırma, uzun okuma dizilemesinin, tek bir testle ve çok daha düşük bir maliyetle, teşhis süresini yıllardan günlere düşürürken, teşhis oranını artırma potansiyeline sahip olduğunu gösteriyor.
The American Journal of Human Genetics dergisinde yayınlanan araştırmaya , UCSC Genomics Institute Biyomoleküler Mühendislik (BME) Profesörü Benedict Paten ve BME Doçenti Karen Miga’nın yanı sıra eski UCSC doktora sonrası araştırmacısı Jean Monlong öncülük etti.
“Nadir hastalıklar, insanların uzun yıllardır teşhis etmekte zorlandıkları bir şey ve eğer teşhis testlerini kolaylaştıran bir dizileme teknolojimiz varsa, bunun çok büyük bir katkı sağlayacağını düşünüyorum ve bu makalede test ettiğimiz şey de buydu,” diyor makalenin ilk yazarı olan UC Santa Cruz BME’de doktora öğrencisi olan Shloka Negi.
“Bugün, genetik dizilemenin tanısal verimi sinir bozucu derecede düşük,” dedi Paten. “Muhtemel nedenlerden biri, klinik uygulamada kullanılan eksik dizileme yöntemleridir. Bu çalışmada, yeni, daha kapsamlı uzun okuma dizilemesinin genetik tanı için yararlı ek bilgiler üretebileceği hipotezini test ediyoruz.
“Kohortumuzda çok sayıda ek potansiyel olarak ilginç genetik varyant ve epigenetik sinyal keşfetmekten heyecan duyduk. Hala erken günler olsa da, bu bilgide büyük bir umut var ve topluluğun bu yeni bilgilerin çoğunu yorumlaması ve tam olarak anlaması zaman alacak.”
Nadir hastalık bulma
Bu çalışmada, tek bir genin bozulması sonucu oluşan nadir görülen monogenik hastalıklara odaklanıldı.
Bilim insanları, genetik hastalıkları, bir genin düzgün çalışmasını engelleyebilecek farklılıklar olan varyantları bulmak için genetik materyallerini tarayarak teşhis eder. Bu varyantları bulmak için tipik yaklaşım, kısa okuma dizilemesi adı verilen bir teknik kullanır; bu teknik, genetik baz çiftlerini (adenin (A), sitozin (C), guanin (G) ve timin (T) kombinasyonlarını) her seferinde yaklaşık 150-250’lik diziler halinde okur.
Ancak kısa okuma dizilemesinin sınırlaması, genomun belirli bölgelerindeki, 250 baz çiftinden çok daha uzun olan baz çiftlerinin desenleri gibi, önemli bilgileri kaçırabilmesidir. Ayrıca, hangi varyantların anneden ve hangilerinin babadan miras alındığını belirleme süreci olan “fazlama” işlemini de gerçekleştiremez.
Bu, klinisyenlerin varyantların kimden miras alındığını keşfetmesine yardımcı olabilir; örneğin, iki varyantın aynı ebeveynden mi, her ebeveynden mi miras alındığını veya hiç miras alınmadığını. Bu, özellikle ebeveyn verileri mevcut olmadığında genetik teşhisler için çok yararlı bir bilgi parçası olabilir.
Buna karşılık, uzun okuma dizilemesi, bilim insanlarının ve klinisyenlerin gen varyasyonu hakkında önemli bilgileri kaçırmasına yol açabilecek boşlukları ortadan kaldırarak, uzun DNA parçalarını aynı anda okuyabilir. Uzun okuma dizilemesi, ayrıca doğrudan fazlama verilerinin yanı sıra, genlerin “açılıp kapanmasına” neden olan ve hastalığa katkıda bulunabilen DNA’daki bir kimyasal süreç olan metilasyon hakkında bilgi sağlar.
Negi, “Uzun okuma dizilemesi belirli durumlarda çok daha iyi olacak ve bunu kanıtlamak için adımlar atıyoruz” dedi.
Yöntemlerde lider
UC Santa Cruz Genomics Institute araştırmacıları, uzun okuma dizilemesinde zengin bir yenilik ve uzmanlık geçmişine sahiptir ve çok çeşitli sağlık araştırma uygulamaları için dizileme ve analizi optimize etmek için aktif olarak yöntemler geliştirmektedir. Araştırmacıların, ilk gerçek anlamda eksiksiz “telomerden telomere” referans genomu gibi başarılar elde etmek için geliştirdikleri tekniklerin çoğu , artık hasta sonuçlarını iyileştirmek için kullanılıyor.
Miga, “Daha önceki bulguları destekleyerek, mevcut eksik ancak yaygın olarak kullanılan genomik referans yerine, tam, sözde ‘telomerden telomere’ referans genomu kullanıldığında, uzun okuma dizilemesinin faydalarının önemli ölçüde arttığını bulduk” dedi.
“İnsanlardaki çeşitli varyasyonları temsil eden referanslar olan pangenomların, yeni uzun okuma dizileme teknolojilerinden daha da fazla fayda sağlayacağını öngörüyoruz.”
Paten ve Miga’nın laboratuvarları, nadir hastalıkları olan 42 hastanın vakaları üzerinde çalışmak için klinisyenlerle ortaklık kurdu; bunlardan bazıları kısa okuma yöntemleri veya diğer özel testler yoluyla teşhis aldı ve bazıları da henüz teşhis edilmedi. Bazı vakalarda, araştırmacılar ebeveyn genetik bilgilerine erişebildiler, ancak diğerlerinde erişemediler.
Hastaların uzun okuma dizilimi, UCSC’de öncülüğü yapılan bir uzun okuma dizileme yöntemi olan nanopore dizileme kullanılarak Miga Laboratuvarı tarafından yürütüldü ve örnek başına yaklaşık 1.000 dolar karşılığında hastaların genomlarının uçtan uca son derece doğru okumaları elde edildi.
Genomik veriler, küçük ve büyük varyantları, faz verilerini ve metilasyon verilerini bulmak için Paten’in laboratuvarında geliştirilen hesaplama yöntemleri kullanılarak analiz edildi ve hepsi Napu boru hattı adı verilen tek bir boru hattı kullanılarak yapıldı . Analiz süreci, bilgisayar işlem hızına bağlı olarak yaklaşık bir gün veya daha kısa sürer ve 100 dolara mal olur.
Vakaları çözmek
Hasta verilerini dizileyip analiz ettikten sonra araştırmacılar, uzun okumaların, kısa okuma dizilemesiyle elde edilebilecek olana kıyasla daha kapsamlı bir veri seti sağladığını buldular.
Uzun okuma dizilemesi, kohorttaki 42 hastanın 11’i için kesin tanı sağladı ve kısa okuma verilerinde bilinen her şeyin yanı sıra ek nadir aday varyantlar, uzun menzilli evreleme ve metilasyon gibi ek bilgileri de tek, uygun maliyetli ve hızlı bir protokolde sağladı.
Teşhis edilen 11 vaka arasında konjenital adrenal hipoplazi (adrenal bezlerinin büyüdüğü ve düzgün çalışmadığı nadir bir durum) vakalarından dördü yer almaktadır. Bu hastalıktan sorumlu gen, genomun özellikle zorlu bir bölgesindedir; kısa okuma dizileme teknolojisiyle karakterize edilemez ve mevcut klinik test zahmetli ve eksiktir.
“Bu vakaları çözmek için, ‘telomer-telomer’ referans genomu gibi yeni yüksek kaliteli montajları entegre eden yeni bir pangenomik araç geliştirdik,” diyor bu projeye Paten’in laboratuvarında doktora sonrası araştırmacı olarak başlayan ve şu anki pozisyonuna Fransa’daki INSERM’de devam eden Monlong.
“Kohortumuzda bu hastalıktan muzdarip dört hastanın patojenik varyantlarını bulup aşamalandırabildiğimizi görmek bizi heyecanlandırdı. Gelecekte, hızlı ve kapsamlı bir klinik test sunabilir. Birçok nadir hastalığın, tarihsel olarak çalışılması zor olan insan genomu bölgelerini içerdiğini biliyoruz, bu nedenle sonuçlarımız yaklaşımımızı uzun süredir duraklamada olan hastalıklara daha fazla genişletmemizi teşvik ediyor.”
Ek olarak, iki vaka cinsiyet gelişimi bozukluklarını içeriyordu, nadir görülen bir Leydig hücre hipoplazisi vakası ise testislerdeki az gelişmiş Leydig hücreleri nedeniyle erkek cinsiyet gelişimini etkiledi. Ek olarak, her biri uzun ve zorlu tanı yolculuklarını temsil eden dört nörogelişimsel bozukluk vakası sonunda çözüldü.
“Uzun okuma dizilemesi, tek bir gende veya net bir fenotipte zorlayıcı varyantlar bulunan çözülememiş vakalar için muhtemelen bir sonraki en iyi testtir,” dedi Negi. “Tek bir tanı testi olarak hizmet edebilir, birden fazla klinik ziyaret ihtiyacını azaltabilir ve yıllar süren bir tanı yolculuğunu saatler meselesine dönüştürebilir.”
Ortalama olarak her hastanın 280 geni ( tek gen mutasyonlarının neden olduğu kalıtsal bozukluklarla bağlantılı olan bazı Mendel hastalığı genleri de dahil ) vardı ve uzun okumalarla benzersiz şekilde kapsanan ve kısa okumalarla tespit edilemeyen önemli protein kodlama bölgeleri vardı.
“Uzun okumaların kilidini açabileceği genomun çok daha fazlası var,” dedi Negi. “Ancak, uzun okumaların ortaya çıkardığı bu yeni bilgiyi tam olarak yorumlayabilmemiz biraz zaman alacak. Bu veriler, kısa okuma analizi ve standart referansa eşleme kullanılarak oluşturulan klinik veri tabanlarımızda yoktu.
“Uzun okumaların, kısa okumaların erişemediği telomer-telomer genomunun yaklaşık %5,8 daha fazlasını açığa çıkardığını gösterdik.”
Gen düzenlenmiş toprak bakterileri, mısır üretimi için üçüncü azot kaynağı sağlayabilir. için yorumlar kapalıBİLİM, GENETİK
Mısır, soya fasulyesinin azot sabitleyici bakterilerle olan ilişkisini kıskandıysa, gen düzenlemedeki ilerlemeler bir gün oyun alanını bile yapabilirdi. Yakın zamanda yapılan bir araştırma, gen düzenlenmiş bakterilerin, erken mısır büyümesi sırasında havadan 35 pound nitrojen eşdeğeri sağlayabildiğini ve bu da mahsulün nitrojenli gübreye olan bağımlılığını azaltabileceğini gösteriyor.
Mısır, soya fasulyesinin azot sabitleyici bakterilerle olan ilişkisini kıskandıysa, gen düzenlemedeki ilerlemeler bir gün oyun alanını bile yapabilirdi. Illinois Üniversitesi Urbana-Champaign’den yakın zamanda yapılan bir araştırma, gen düzenlenmiş bakterilerin erken mısır büyümesi sırasında havadan 35 pound nitrojen eşdeğeri sağlayabildiğini ve bu da mahsulün nitrojenli gübreye olan bağımlılığını azaltabileceğini gösteriyor.
“Tüm sentetik nitrojeni değiştirmek kesinlikle bir şey olurdu. Belki bundan 100 yıl sonra, bu hedefe yaklaşmak için mikropları ve genetik ince ayarları bulmuş olacağız, ancak bu mikroplar henüz orada değil. Bununla birlikte, bir yerden başlamalıyız ve bu çalışma mısır için azot fiksasyonunun potansiyele sahip olduğunu gösteriyor “diyor Illinois’deki Tarım, Tüketici ve Çevre Bilimleri Koleji’nin bir parçası olan Mahsul Bilimleri Bölümü’nde araştırma görevlisi olan ortak yazar Connor Sible.
Sible ve ortak yazarları, atmosferik nitrojeni bitkide bulunan formlara dönüştürebilen, sırasıyla bir veya iki toprak bakterisi türü içeren PROVEN ve PROVEN® 40 adlı Pivot Bio’nun ürünlerini test etti. Düzenlenmiş versiyonlar, nitrojen fiksasyonunda yer alan önemli bir genin aktivitesini artırarak bitkiler için daha fazlasını kullanılabilir hale getirir. Dikim sırasında uygulandığında, bakteriler bitki köklerini kolonize ederek besini en çok ihtiyaç duyulan yere ulaştırır.
Şirket, biyolojik olarak sabitlenmiş nitrojenin potansiyel olarak dönüm başına 40 pound’a kadar gübre azotunun yerini alabileceğini iddia ediyor.
“Bu iddiayı destekleyecek hakemli yayınlanmış veri eksikliği var. Çalışmayı Illinois’de doktora öğrencisi olarak tamamlayan Logan Woodward, nitrojen replasman değerlerinin büyüklüğünü ve büyüme döngüsünde ne zaman ek nitrojen biriktiğini tahmin eden bir araştırma da yoktur “dedi. “Amacımız bu bilgi boşluklarını doldurmaktı.”
Araştırmacılar, ürünleri, dönüm başına 0, 40, 80, 120 veya 200 pound azotlu gübre de dahil olmak üzere mısır için standart agronomik uygulamaları kullanarak üç tarla mevsimi boyunca ekimde uyguladılar. Daha sonra V8 aşamasında (sekiz tam yakalı yaprak) ve R1’de (ipek çıkışı) bitki dokularındaki azotu ve her mevsimin sonunda tahıl verimini ölçtüler. Bitki ve toprak stabil izotopik azotun seyreltilmesi, aşılanan arazilerde ek azot alımının atmosferden olduğunu ve toprak ve gübre tedarikini desteklediğini gösterdi.
Analiz, tüm azotlu gübre oranlarında, aşılayıcının mısır vejetatif büyümesini, azot birikimini, çekirdek sayısını ve verimi dönüm başına ortalama 2 kile artırdığını gösterdi. Orta azot oranlarında, verim dönüm başına 4 kile arttı. Bu, dönüm başına gübre başına 10-35 pound azota eşdeğerdi.
“Genel verim tepkisi olumluydu, ancak mütevazıydı. Erken büyüme sırasında 35 pound gübre eşdeğeri, sezon sonunda yaklaşık 10’a düştü, “dedi mahsul bilimleri profesörü kıdemli çalışma yazarı Fred Below. “Açıkçası, hala gübrelemeye ihtiyaç var. Mutlu ve sağlıklı bir bitki oluşturmak için yeterli nitrojene ihtiyacınız vardır, çünkü sağlıklı bir bitki daha sonra mikropları beslemek için gereken kök şekerlerini üretebilir. Azot olmadan bitki kendini veya aşılanmış mikropları destekleyemez, bu nedenle bir miktar gübre azotunun yokluğunda etkinlik oldukça azalır.
Genetik mutasyonun Huntington hastalığına yol açmasının şaşırtıcı yolu, hastalığa ilişkin anlayışı değiştiriyor için yorumlar kapalıGENETİK
MIT ve Harvard’ın Broad Enstitüsü, Harvard Tıp Fakültesi ve McLean Hastanesi’ndeki bilim insanları, Huntington hastalığına neden olduğu bilinen kalıtsal genetik mutasyonun beyin hücrelerinin ölümüne yol açtığı şaşırtıcı bir mekanizma keşfettiler. Bulgular, ölümcül nörodejeneratif bozukluk hakkındaki anlayışı değiştiriyor ve onu geciktirmenin veya hatta önlemenin olası yollarını öneriyor.
Cell’de yayınlanan bir çalışma, kalıtsal mutasyonun hücrelere zarar vermediğini ortaya koyuyor. Aksine, mutasyon onlarca yıl zararsız ama yavaş yavaş hücreyi hızla öldüren oldukça toksik bir forma dönüşüyor.
Huntington mutasyonu, HTT genindeki bir DNA bölümünü içerir; bu bölümde, üç harfli bir DNA dizisi olan “CAG”, hastalığı olmayan kişilerde miras alınan 15-35 tekrarın aksine en az 40 kez tekrarlanır. Araştırmacılar, 40 veya daha fazla CAG tekrarı olan DNA yollarının yüzlerce tekrar uzunluğunda olana kadar büyüdüğünü buldular.
Bu tür “somatik genişleme” yalnızca Huntington hastalığında daha sonra ölen belirli beyin hücresi tiplerinde meydana gelir. Bir hücrenin DNA genişlemesi yalnızca CAG’lerin eşik sayısına ulaştığında (yaklaşık 150) hücre hastalanır ve sonra ölür. Bu tür birçok hücrenin kümülatif ölümü Huntington hastalığının semptomlarına yol açar.
Çalışma, HTT proteininin ifadesini azaltmayı amaçlayan aday Huntington ilaçlarının klinik deneylerde neden zorluk çektiğine dair olası bir açıklama sunuyor: Herhangi bir anda proteinin toksik versiyonuna sahip çok az hücre var, bu nedenle tedaviler çoğu hücrede terapötik bir etkiye sahip olmayabilir.
Araştırma ayrıca farklı bir tedavi stratejisini de gündeme getiriyor: HTT genindeki CAG tekrar genişlemesini durdurmak veya yavaşlatmak, çok daha fazla sayıda hücrede toksisiteyi erteleyebilir ve hastalığın başlangıcını geciktirebilir, hatta önleyebilir.
Çalışmanın ortak yazarlarından genetikçi, sinir bilimci ve aynı zamanda araştırmacı Steve McCarroll, “Bu deneyler, Huntington hastalığının nasıl geliştiğine dair düşünce şeklimizi değiştirdi” dedi.
McCarroll, Broad’daki Stanley Psikiyatri Araştırma Merkezi’nde enstitü üyesi ve genomik nörobiyoloji direktörü, Harvard Tıp Fakültesi’nde Dorothy ve Milton Flier Biyomedikal Bilim ve Genetik Profesörü ve Howard Hughes Tıp Enstitüsü’nde araştırmacıdır.
“Bu, bir mutasyonun bir hastalığa nasıl yol açtığı konusunda gerçekten farklı bir düşünme biçimi ve bunun Huntington hastalığının ötesinde DNA tekrar bozukluklarında da uygulanabileceğini düşünüyoruz.”
Harvard Tıp Fakültesi ve McLean Hastanesi’nde psikiyatri doçenti ve Mass General Brigham sağlık sisteminin bir üyesi olan kıdemli ortak yazar Sabina Berretta, “Çalışmamızın amacı, hepimizin yaptığı şey, hastalığın neden olduğu acıyı hafifletmektir” dedi.
Ayrıca McLean Hastanesi’ndeki bir NIH NeuroBioBank merkezi olan Harvard Beyin Dokusu Kaynak Merkezi’nin (HBTRC) direktörüdür. “Bu çalışma ve bilgilendirdiği çalışma etkili olabilir ve kısa vadede acıyı hafifletmede büyük bir fark yaratabilir.”
McCarroll’un grubundan kadrolu bilim insanı Bob Handsaker, kıdemli baş yazılım mühendisi Seva Kashin ve eski araştırma görevlisi Nora Reed, çalışmanın ortak birinci yazarlarıdır.
Açık sorular
Huntington hastalığı, beynin derinliklerinde hareketten, birçok bilişsel işlevden ve motivasyondan sorumlu bir yapı olan striatumda bulunan striatal projeksiyon nöronları adı verilen bir hücre popülasyonunu öldürür.
Bu hücrelerin büyük bir kısmı öldüğünde, hastalar kollarında, bacaklarında ve yüzlerinde istemsiz hareketler geliştirir ve birçok hastada bilişsel sorunlar da gelişir. Bu semptomlar genellikle orta yaşta başlar ve daha sonra 10 ila 20 yıl içinde daha şiddetli bilişsel sorunlara ve hareket etme veya yutma zorluğuna ilerler.
Tekrarların süresi ne kadar uzun olursa, kişi semptomlar ilk ortaya çıktığında o kadar genç olma eğilimindedir. Tekrarlanan CAG’lerin yolunun da zamanla genişlediği ve farklı dokularda çeşitli uzunluklara yol açtığı gösterilmiştir.
Ancak altta yatan biyolojik sorular her zaman devam ediyordu: HTT mutasyonu nasıl toksiktir? Vücudun hemen hemen her hücresinde görülen HTT proteini neden sadece bazı beyin hücrelerini öldürüyor da diğerlerini öldürmüyor? Ve mutasyonla doğan ve proteini yaşam boyunca ifade eden hastalar neden sadece orta yaşta, onlarca yıllık görünür sağlık durumundan sonra semptomlar geliştiriyor?
CAG tekrar uzunluğunun doğrudan biyolojik etkilerini anlamaya çalışan araştırmacılar, yalnızca gen ifadesini ve tek hücrelerin kimliğini değil, aynı zamanda her hücrenin içindeki DNA tekrar yollarının uzunluğunu da belirlemelerine yardımcı olmak için tek hücreli RNA dizilemesini uyarladılar.
“Bu tekrarların nöronlarda genişlediği biliniyor,” dedi Kashin. “Ancak belirli bir hücreyi alıp hem CAG uzunluğunu hem de transkripsiyonel profili ölçme yeteneği, gerçekten güçlü bir analize izin veren gerçekten önemli bir temeldir.”
Araştırmacılar, Huntington hastalığı olan 53 kişi ve hastalığı olmayan 50 kişiden alınan beyin dokularını HBTRC tarafından toplayıp sakladılar.
500.000’den fazla tek hücreyi analiz ettiler ve hastalığı olan kişilerin çoğu hücre tipinin, miras aldıkları CAG tekrarının esasen aynısına sahip olduğunu buldular. Ancak striatal projeksiyon nöronları (hastalıkta ölen birincil striatal hücreler) CAG tekrar yollarını büyük ölçüde genişletmişti.
İnsan beyin dokusu üzerine daha önce yapılan araştırmaların çoğu, 100’den az tekrar içeren CAG tekrar yollarına odaklanmıştı; ancak yeni çalışma, bazı nöronların 800’e kadar CAG’ye sahip olduğunu gösterdi ve bu, Glasgow Üniversitesi’nden Peggy Shelbourne’un 20 yıl önce yaptığı keşfi doğruladı.
En şaşırtıcı olanı, araştırma ekibinin DNA tekrarının 40’tan 150 CAG’a genişlemesinin nöronların sağlığı üzerinde görünür bir etkisi olmadığını bulmasıdır. Ancak tekrarları 150 CAG’ı aşan nöronlar büyük ölçüde bozulmuş gen ifadesi, kritik genlerin ifadesini kaybetme ve ardından ölme gösterdi.
McCarroll’un ekibi ayrıca, striatal projeksiyon nöronlarında CAG tekrar genişlemesinin hızını ve zamanlamasını tahmin etmek için deneysel verilerin bilgisayar modellemesini kullandı.
CAG tekrar yollarının başlangıçta yavaş büyüdüğünü, yaşamın ilk iki on yılında yılda bir kereden az genişlediğini buldular. Ancak bir hücrenin tekrar yolu yaklaşık 80 CAG’ye ulaştığında -genellikle birkaç on yıl sonra- genişleme hızı önemli ölçüde hızlanır ve sadece birkaç yıl içinde 150 CAG’ye genişler.
Hücre daha sonra sadece birkaç ay sonra ölür. Bu, bir nöronun hayatının %95’inden fazlasını zararsız bir HTT geniyle geçirdiği anlamına gelir. Dahası, farklı hücrelerdeki CAG tekrar yolları bu toksisite eşiğini farklı zamanlarda geçtiği için, hücreler bir grup olarak uzun bir süre boyunca yavaşça kaybolur, semptomlar ortaya çıkmadan yaklaşık 20 yıl önce başlar ve semptomlar başladıkça daha hızlı bir şekilde kaybolur.
“Bu çalışmaya başlamadan önce Huntington hastalığı hakkında çok şey biliniyordu, ancak kolektif anlayışımızda boşluklar ve tutarsızlıklar vardı,” dedi Handsaker. “Onlarca yıl boyunca tek tek nöronlarda ortaya çıkan patolojinin tam gidişatını bir araya getirebildik ve bu bize terapötik olarak müdahale edebileceğimiz potansiyel olarak birçok farklı zaman noktası sağlıyor.”
Huntington hastalarının beyin dokularını analiz etmek çalışma için kritik önem taşıyordu. Berretta, “Çok zor bir şeyi yapmayı seçen ailelere minnettarız,” dedi. “Bu, kalıcı ve birçok başka insana fayda sağlayacak bir bilgi mirası bırakan birçok beyin bağışçısının fedakarlığı olmadan mümkün olmazdı.”
Terapötik olanaklar
McCarroll’un ekibi, HTT proteinini hedeflemek yerine, tamamlayıcı veya potansiyel olarak daha iyi bir tedavi yaklaşımının, DNA tekrar genişlemesini yavaşlatmak veya durdurmak olabileceğini, bunun da hastalığı geciktirmeye veya hatta önlemeye yardımcı olabileceğini öne sürüyor.
Massachusetts General Hastanesi’nden Vanessa Wheeler ve Ricardo Mouro Pinto’nun çalışmaları da dahil olmak üzere Huntington’a dair daha önce yapılan genetik çalışmalar , bu yayılmayı yavaşlatmanın olası yollarına işaret ediyor.
Çalışmalar, DNA’yı koruma ve onarmada rol oynayan hücresel proteinlerin bazen DNA tekrar yollarının kararlılığını zayıflattığını gösterdi. Örneğin, MSH3 proteini normalde hücrenin DNA’sını olası mutasyonlara karşı izlemesine yardımcı olur, ancak ekstra CAG’ler tarafından oluşturulan DNA’daki halkalar bu proteini CAG tekrarını genişletmeye yönlendirebilir.
Uluslararası bir insan genetikçileri ekibi, bu DNA onarım proteinlerini kodlayan genlerdeki yaygın genetik varyasyonların Huntington hastalarında semptomların başlangıcını hızlandırabileceğini veya geciktirebileceğini buldu . McCarroll, bu bulguların ekibinin tek hücrelerde CAG tekrarını ölçmenin yollarını geliştirmeye odaklanmasına doğrudan ilham verdiğini söylüyor.
Moleküler bir terapi ile belirli DNA bakım süreçlerinin yavaşlatılmasının, daha az hataya neden olan diğer DNA onarım mekanizmalarının bu döngüleri çözmesine izin vererek DNA tekrar genişlemesini yavaşlatabileceğini de ekliyor.
Bu arada araştırmacılar, 150 CAG’den daha uzun DNA tekrar yollarının nöronal bozulmaya ve ölüme nasıl yol açtığını ve tekrarların neden bazı nöron türlerinde diğerlerinden daha fazla genişlediğini anlamak için çalışıyorlar. Ayrıca, hastalarda DNA tekrarlarını ve geç başlangıcı içeren diğer genetik bozukluklardaki DNA tekrar genişlemesi ile hücresel değişiklikler arasındaki bağlantıyı anlamak için DNA tekrar profiliyle birlikte tek hücreli RNA dizilemesinin benzer bir kombinasyonunu kullanıyorlar.
Kırılgan X sendromu ve miyotonik distrofi de dahil olmak üzere 50’den fazla insan beyin rahatsızlığı, çeşitli genlerdeki DNA tekrarlarının genişlemesinden kaynaklanmaktadır.
McCarroll, “DNA tekrarlarının genişlemesini yavaşlatan tedavilere ulaşmak için birçok insanın çok fazla bilimsel çalışma yapması gerekecek,” dedi. “Ancak bunun merkezi hastalık yönlendirme süreci olarak anlaşılmasının derin bir odaklanmaya ve yeni seçeneklere yol açacağını umuyoruz.”