Soyut
Protein-DNA etkileşimlerinin (PDI’ler) doğru bir şekilde ölçülmesi, biyolojik süreçleri anlamak ve ilaç tasarımını kolaylaştırmak için kritik öneme sahiptir. Ancak, nükleik asitlerin doğasında bulunan esneklik, deneysel olarak belirlenmiş PDI kompleks yapılarının kullanılabilirliğini sınırlayarak güvenilir puanlama fonksiyonlarının (SF’ler) eğitilmesinde önemli bir zorluk oluşturmaktadır. Bu sorunu çözmek için, PDI tahmini için yeni bir derin öğrenme tabanlı SF olan PDIScore’u geliştirdik. PDIScore, nükleotit esnekliğini yakalamak için kapsamlı bir grafik gösterimi kullanır, büyük etkileşim arayüzlerini yönetmek için BigBird doğrusal küresel dikkat özelliğine sahip ölçeklenebilir bir GraphGPS mimarisi kullanır ve kalıntı-nükleotit mesafe dağılımlarını modellemek için Karışım Yoğunluk Ağları’ndan (MDN’ler) yararlanır. PDIScore, yaklaşık 7000 protein-nükleik asit kompleks yapısından oluşan kendi kendine toplanan bir veri kümesi üzerinde eğitilmiş ve tarama, yerleştirme ve sıralama yeteneklerini değerlendirmek için üç titiz test kümesi üzerinde doğrulanmıştır. Sonuçlar, PDIScore’un mevcut yöntemlerden önemli ölçüde daha iyi performans gösterdiğini göstermiştir: tarama setinde en iyi tarama gücüne (örneğin, AlphaFold3 yapıları kullanılarak EF %1 = 14,13, AUROC = 0,82), yerleştirme setinde en yüksek yerleştirme başarı oranına (%48,94 ilk 1) ve sıralama setinde üstün sıralama yeteneğine (PCC = 0,50) ulaşmıştır. Vaka çalışmaları, PDIScore’un biyolojik mekanizmaları (örneğin, adenovirüs transkripsiyonu, SOCS1 regülasyonu) açıklama yeteneğini ve önemli etkileşim bölgelerini belirlemek için nükleotid düzeyinde yorumlanabilirliğini göstermiştir. PDIScore, PDI ile ilgili araştırmaları ve tedavi tasarımını ilerletmek için önemli bir potansiyele sahip, sağlam ve genelleştirilebilir bir araçtır.
giriiş
Protein-DNA etkileşimleri (PDI’ler), DNA replikasyonu, RNA transkripsiyonu, gen onarımı ve gen düzenlemesi dahil olmak üzere birçok biyolojik süreç için temeldir [ 1 , 2 ]. PDI’ler ayrıca iltihaplanma, kanser ve Alzheimer hastalığı gibi çeşitli hastalıklarla da ilişkilidir [ 3 , 4 , 5 ]. Bu etkileşimleri anlamak, yaşam mekanizmaları, hastalık yolları ve ilaç keşfi hakkında değerli bilgiler sunar [ 6 , 7 , 8 ]. İlk ikisi tipik olarak, ilişkili genlerin transkripsiyonunu düzenlemek için DNA dizilerini özel olarak tanıyan TATA-kutusu bağlayıcı protein (TBP) ve erken büyüme tepkisi proteini 1 (Egr1) gibi transkripsiyon faktörleriyle (TF’ler) ilişkilidir [ 9 , 10 ]. Örneğin, adenovirüs replikasyon döngüsü sırasında TBP, adenovirüs majör geç promotörünün (AdMLP) TATA kutusuna bağlanır ve böylece adenovirüs RNA’sının transkripsiyonunu aktive eder [ 11 ]. Hastalık yollarıyla ilgili olarak, Egr1, sitokin sinyallemesinin baskılayıcısı-1’in (SOCS1) transkripsiyonunu düzenler; bu bozukluğun aşırı iltihaplanma, otoimmün durumlar ve maligniteler dahil olmak üzere bağışıklık kusurlarına yol açması mümkündür [ 12 , 13 ]. PDI ile ilişkili ilaçlara gelince, DNA aptameri [ 14 , 15 ] gibi protein hedefli terapiler ve siklik peptitler [ 16 ] gibi DNA hedefli ilaçlar vardır . Dahası, proteinlere karşı yüksek afinite ve özgüllük için tasarlanmış sentetik DNA’lar, spesifik proteinlerin rolünü açıklamak için ilaç, tanı aracı ve antagonist olarak geliştirilmektedir [ 17 ].
Yüksek verimli çalışmalarda PDI’ların kantitatif analizi, sentetik DNA’nın rasyonel tasarımı için esastır. Bununla birlikte, elektroforetik hareketlilik kayması testi [ 18 ], izotermal titrasyon kalorimetrisi [ 19 ] ve yüzey plazmon rezonansı [ 20 ] gibi deneysel yöntemler, protein-DNA bağlanma afinitesini hızlı ve doğru bir şekilde ölçmede önemli zorluklarla karşı karşıyadır. Bu zorluklar, kısa DNA’nın yapısal son etkilerinden ve uzun DNA’nın spesifik olmayan bağlanma bölgelerinin bolluğundan kaynaklanmaktadır [ 18 ]. Dahası, bu yöntemler, özellikle yüksek verimli deneylere uygulandığında, genellikle zaman alıcı ve maliyetlidir [ 21 ]. Bu nedenle, hem dizi tabanlı hem de yapı tabanlı yöntemler dahil olmak üzere, protein-DNA bağlanmasını hızla tahmin edebilen hesaplamalı yöntemlere geçici bir ihtiyaç vardır. Nükleik asitlerin içsel esnekliği ve çeşitli konformasyonları, dizi tabanlı yöntemler kullanılarak doğru bağlanma afinitesi tahminlerini zorlaştırır. Sonuç olarak, yapıya duyarlı yöntemlerin genellikle dizi tabanlı yöntemlere kıyasla daha yüksek tahmin doğruluğu sağlaması beklenir [ 22 ].
Protein-DNA bağlanma afinitesini tahmin etmek için bir dizi yapı tabanlı hesaplamalı puanlama fonksiyonu (SF) geliştirilmiştir [ 21 , 23 , 24 , 25 , 26 , 27 , 28 ]. Ancak, nükleik asitlerin içsel esnekliği, bilinen bağlanma afinitelerine sahip deneysel olarak belirlenmiş yapıların kıtlığına yol açar ve bu da genellikle 500’den az girdi içeren veri kümelerine dayanan mevcut yöntemler için önemli bir zorluk oluşturur [ 29 ]. MM/GBSA [ 25 ], FoldX [ 23 , 24 ] ve PyRosetta’daki ref2015 ve dna_gb [ 26 ] gibi geleneksel yaklaşımlar , genellikle PDI’ları birden fazla ampirik enerji teriminin ağırlıklı toplamı olarak ölçer. Makine öğrenimi (ML) algoritmalarının ortaya çıkışı, PreDBA [ 27 ], emPDBA [ 28 ] ve SAMPDI-3D [ 21 ] tarafından temsil edilen, doğrudan verilerden öğrenebilen bir ML tabanlı SF (MLSF) havuzunun geliştirilmesini kolaylaştırmıştır. Bu yöntemler, dahili veri kümelerinde etkileyici bir performans gösterse de, bu veri kümelerinin sınırlı boyutu ve harici doğrulama veya sistematik değerlendirmenin eksikliği, güvenilirliklerini tehlikeye atabilir [ 29 ]. Örneğin, SAMPDI-3D’nin en büyük veri kümesi, eğitim için 463 mutant içeriyordu [ 29 ]. Değerlendirmemize göre, bu SF’lerin çoğu, bilinen bir dizi bağlayıcıyı sıralamakta zorlanıyor (sıralama gücü) ve bazıları aktif bağlayıcıları bağlayıcı olmayanlardan veya zayıf aktif bağlayıcılardan (tarama gücü) bile neredeyse ayırt edemiyor.
Yerel benzeri bağlanma pozlarını yanlış olanlardan ayırt etme yeteneği (yerleştirme gücü), bir SF’yi değerlendirmek için önemli bir kriterdir. Yukarıda belirtilen yöntemler puanlama için karmaşık yapılara ihtiyaç duyar, ancak nükleik asitlerin esnekliği genellikle kristal yapıların bulunabilirliğini sınırlar ve protein-DNA kompleks yapılarını tahmin etmek için yerleştirme tekniklerinin kullanımını gerektirir. Temsili yerleştirme yöntemleri arasında HDOCK [ 30 ], PyDockDNA [ 31 ] ve HADDOCK [ 32 , 33 ] bulunur. Bu arada, AlphaFold [ 34 ] gibi yapay zeka (AI) tabanlı yapı tahmin yöntemleri, protein yapı tahmininde dikkate değer bir başarı göstermiştir ve en son sürümü olan AlphaFold3 [ 35 ], protein-DNA komplekslerini doğrudan dizilerden tahmin edebilir. Bu yöntemler, birden fazla yerleştirme pozunu sıralamak ve filtrelemek için dahili SF’lere sahiptir. SF’lerin yerel benzeri bağlanma pozlarını yanlış olanlardan ayırt etme yeteneği, doğru yapı tahmini ve sonraki bağlanma afinitesi tahmini için çok önemlidir.
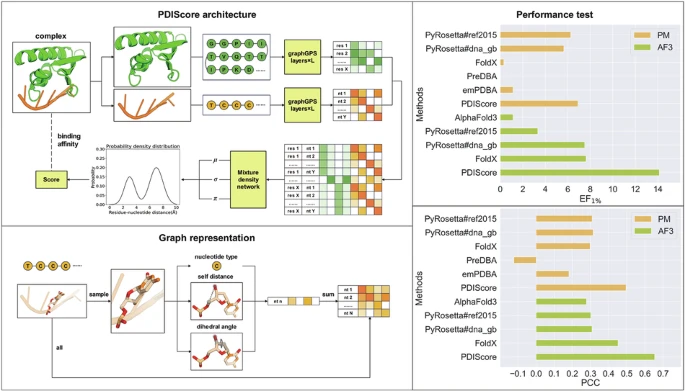
Daha güvenilir bir SF geliştirmek için, Protein Veri Bankası’ndan (PDB) [ 36 ] alınan yaklaşık 7000 protein-nükleik asit kompleks yapısından oluşan kapsamlı bir veri seti oluşturduk. Bildiğimiz kadarıyla, bu kadar büyük ölçekli bir yapı veri seti daha önce hiç rapor edilmemiştir ve bu, tahmin modellerinin eğitimi için öncü bir kaynak olarak kullanılabilir ve böylece PDI’ların incelenmesini ilerletebilir. Mevcut PDI SF’lerini kapsamlı bir şekilde değerlendirmek için, (1) aynı araştırma grubu tarafından aynı deneysel yöntem kullanılarak belirlenen bağlanma afinitelerine sahip 5 sistem ve toplam ~28000 protein-DNA kompleksi içeren bir tarama seti [ 37 ], (2) 47 bağlı olmayan protein-DNA kompleks yapısı içeren yayınlanmış bir yerleştirme kıyaslaması [ 38 ] ve (3) hem protein mutasyonlarını hem de DNA mutasyonlarını kapsayan ölçülen bağlanma afinitelerine sahip 9 sistem ve ~200 protein-DNA kompleksi içeren bir sıralama seti [ 21 , 39 , 40 , 41 , 42 ] olmak üzere üç doğrulama seti de derledik. Tarama setindeki her bir ligandın bilinen bağlanma afiniteleri nedeniyle, sıralama gücünü değerlendirmek için de kullanılabilir. Bu veri setlerine dayanarak, PDI tahmini için PDIScore adlı derin öğrenme (DL) tabanlı bir yaklaşım sunduk. Model mimarimiz dört ana modülden oluşmaktadır: grafik gösterimi, özellik çıkarma, birleştirme ve karışım yoğunluk ağı (MDN). Nükleik asitlerin proteinlere kıyasla daha yüksek konformasyonel esnekliği göz önüne alındığında, grafik gösterimine daha geniş bir yapısal tanımlayıcı dizisi (11 atom mesafesi ve 20 dihedral açı) kullandık. Özellik çıkarma için, önceki çalışmalarımızda kullanılan tam bağlı grafik dönüştürücüsünün (GT) yerine BigBird [ 44 ] tarafından sağlanan doğrusal küresel dikkat ile genel, güçlü, ölçeklenebilir (GPS) grafiği [ 43 ] kullandık [ 45 , 46 ]. Bu özelliklendirme stratejisi, birkaç bin düğüme sahip grafiklere ölçeklenebilir ve bu da onu bu çalışma için özellikle uygun hale getirir. Protein-küçük molekül etkileşimleriyle karşılaştırıldığında, PDI’lar genellikle daha büyük temas alanlarına sahiptir ve bu, özellik çıkarma sırasında daha fazla düğüm ve kenara sahip daha büyük grafiklerde yansıtılır. MDN modülleri, her kalıntı ile her nükleik asit arasındaki mesafenin olasılık yoğunluk dağılımını öğrenmek için kullanılır. Genel olarak, PDIScore yapı veri kümesi üzerinde eğitilmiş ve tarama, yerleştirme ve sıralama kümelerinde titizlikle test edilmiştir. Bu sayede, deneysel ölçümlerle daha iyi bir korelasyon elde etme ve doğal yapıları tuzak yapılardan ayırt etme becerisini kanıtlamıştır. PDIScore ayrıca, AlphaFold3 için bir yeniden puanlama aracı olarak kullanışlılığını da kanıtlayarak, PDI ile ilgili ilaç tasarımına değerli destek sunmaktadır.
Malzemeler ve yöntemler
Veri seti hazırlama
Bu çalışmada yapı veri seti, tarama seti, yerleştirme seti ve sıralama seti olmak üzere toplam dört veri seti oluşturulmuştur. Yapı veri seti model eğitimi için kullanılmış olup, dağılımı Şekil S1’de gösterilmiştir . Kalan üç set ise model değerlendirmesi için test setleri olarak kullanılmış olup, Şekil S2 ve Tablo S1’de ayrıntılı olarak açıklanmıştır .
Yapı veri seti, PDB’den (16 Ekim 2023’ten önce) alınan 7108 protein-nükleik asit kompleks yapısından oluşuyordu. Bunlar arasında, 532 protein-DNA kompleksi, modeli ince ayarlamak için PDBbind veritabanından (v2020) [ 47 ] alınan afinite verileriyle etiketlendi ve afinite veri setini oluşturdu. Şekil S1’de gösterildiği gibi , yapı veri setinin dağılımı afinite veri setinin dağılımına oldukça benziyordu. Yapı veri setindeki komplekslerin yaklaşık %86’sı ve afinite veri setindeki komplekslerin yaklaşık %97’si 60’tan az nükleotit içeriyordu. Her iki veri seti de aynı en yaygın dört protein türünü paylaşıyordu: transferaz, transkripsiyon, DNA bağlayıcı protein ve hidrolaz.
Tarama seti, DNA ile bağlı vahşi tip kristal yapıları belirlenmiş olan TBP, Ets1, Egr1, Max ve GR dahil olmak üzere beş protein hedefi için ~28.000 protein-DNA kompleksi içeriyordu (PDB girişleri: sırasıyla 1QNE, 2NNY, 1P47, 1AN2 ve 1R4R). Her bir spesifik protein hedefi için, eşit uzunluktaki DNA dizilerinin bağlanma afiniteleri aynı araştırma grubu tarafından aynı biyolojik deney kullanılarak ölçüldü. Kullanılamayan kristal yapılara sahip mutantlar, ilgili vahşi tipten nokta mutasyonu ile elde edildi veya AlphaFold3 ile tahmin edildi. Nokta mutasyonu için, referans kristal yapıları önce DNA dizisi bilgilerine dayalı olarak Chimera [ 48 ] kullanılarak mutasyona uğratıldı, ardından Rosetta [ 49 ] kullanılarak tüm sistemin en aza indirilmesi sağlandı . Alphafold3 için, DNA ve protein dizileri, ilgili yapıları tahmin etmek için girdi olarak kullanıldı. AlphaFold3’e gömülü SF ile adil bir karşılaştırma sağlamak için, tahmin edilen yapılar enerji minimizasyonu ile işlenmedi. Deneysel bağlanma verilerine göre sıralanan en üstteki %1’lik DNA’lar, belirli bir protein için aktif ligandlar olarak kabul edildi. Diğerleri ise tuzak olarak kabul edildi. Bu setin temel amacı, yaklaşımın tüm kütüphanedeki bu ligandları zenginleştirip zenginleştiremeyeceğini test etmekti. Ayrıca, Egr1 ve Max arasındaki her DNA için bağlanma tercihi mevcuttu ve bu tercihler, yaklaşımın bu tercihleri yeniden üretip üretemeyeceğini tahmin etmek için küçük ölçekli bir ters tarama testi olarak kullanılabilirdi.
Yerleştirme seti, 47 bağlı olmayan-bağlı olmayan test vakası içeren yayınlanmış, yedekli olmayan bir protein-DNA yerleştirme kıyaslamasından doğrudan alındı. Bağlı olmayan yapı, bağlanma partnerlerinin yokluğunda veya farklı bir kompleks içinde var olan bir konformasyon olarak tanımlandı. Bağlı olmayan DNA yapıları, 3DNA [ 50 ] programı kullanılarak oluşturuldu ve bağlı olmayan protein yapıları X-ışını veya NMR ile belirlendi. Kıyaslama seti, dizileri yapı veri setindeki dizilere %40’tan fazla benzeyen kompleksleri kaldırarak MMseqs2 [ 51 ] programı kullanılarak yedekliliği azaltıldı . Bu işlemin ardından, yedekli olmayan veri setini oluşturan 41 kompleks kaldı.
Sıralama kümesi, protein ve DNA mutasyonlarını kapsayan farklı mutasyonlardan kaynaklanan bağlanma serbest enerjisindeki (ΔΔG) değişiklikleri yakalamak için her yaklaşımın kabiliyetini değerlendirmek için kullanıldı. Her biri 15’ten fazla mutasyon ΔΔG değeri içeren SAMPDI-3D’nin test kümelerinden sistemler seçtik. Adil bir karşılaştırma sağlamak ve ek veri sağlamak için, SAMPDI-3D’nin eğitim kümelerinde tamamen veya kısmen bulunmayan sistemleri de belirledik, böylece çözülmüş DNA’ya bağlı kristal yapılara sahip CEBPB, MafB, ELK1, ETV5, ERG, Tral, CAP, PadR ve c-Fos-c-Jun dahil olmak üzere dokuz protein bıraktık (PDB girişleri: sırasıyla 1GU4, 2WTY, 1DUX, 4UNO, 4IRI, 2A0I, 1RUN, 5×11 ve 1FOS). İlk beş protein DNA mutasyonlarını içerirken, kalan dört protein protein mutasyonlarını içeriyordu. Yukarıda belirtildiği gibi bilinmeyen yapılar nokta mutasyonu veya AlphaFold3 ile de tahmin edilmiştir.
Grafik gösterimleri
PDIScore’un model mimarisi , grafik gösterimi, özellik çıkarma, birleştirme ve MDN modüllerinden oluşan Şekil 1’de gösterilmiştir . Proteinlerin grafik gösterimleri, protein-küçük molekül etkileşimlerini tahmin etmede etkili olduğunu gösteren önceki çalışmamız RTMScore’dan referans alınmıştır [ 46 ]. Ko-kristalize DNA’nın etrafında 10,0 Å yarıçapında bulunan kalıntılar bağlanma cebi olarak tanımlanırken, diğer kalıntılar bağlanmayla ilgisiz kabul edilerek yapıdan uzaklaştırılmıştır. Daha sonra her bir cep, yönlendirilmemiş bir grafik olarak temsil edilmiştir ( ), düğümler bir cepteki kalıntıları ve kenarlar minimum 10,0 Å’den daha az mesafeye sahip herhangi iki kalıntı arasındaki etkileşimleri temsil etmektedir. Düğümlerin özellikleri, önceki çalışmamızda etkili olduğu gösterilen kalıntı seviyesine dayanmaktadır [ 46 ]. Kenarlar için, proteinin ikincil ve üçüncül yapılarını korumada önemli rol oynayan kovalent olmayan etkileşimleri dikkate aldık. Protein grafiğinin özellikleri Tablo 1’de özetlenmiştir . Özellikle, düğüm özellikleri arasında amino asit türü, öz mesafeler ve her kalıntı için dihedral açılar; kenar özellikleri arasında ise bağlanma durumları, CA-CA mesafeleri, merkezler arası mesafeler ve herhangi iki kalıntı arasındaki maksimum ve minimum mesafeler yer almaktadır.
Kaynak: https://link.springer.com/article/10.1038/s41401-025-01688-3