Terapötikler için etkili mRNA dizileri tasarlamak zorlu bir görev olmaya devam etmektedir. Protein tasarımındaki başarılardan ilham alan dil modelleri (LM’ler) artık RNA’ya da uygulanıyor, ancak kapsamlı eğitim verilerinin eksikliği ilerlemeyi genellikle engelliyor. Mevcut modeller genellikle UTR veya CDS bölgeleriyle sınırlı olduğundan, tam mRNA dizileri için uygulamaları kısıtlanıyor. Mevcut en büyük mRNA veri kümesi üzerinde önceden eğitilmiş, sağlam ve hepsi bir arada bir mRNA tasarımcısı olan mRNABERT’i tanıtıyoruz. Performansı artırmak için, protein dizilerinden gelen anlamsal bilgileri entegre etmek üzere çapraz-modaliteli karşılaştırmalı bir öğrenme çerçevesine sahip ikili bir belirteçleme şeması öneriyoruz. Kapsamlı bir kıyaslamada, mRNABERT, 5′ UTR ve CDS tasarımı, RNA bağlayıcı protein (RBP) bölgesi tahmini ve tam uzunlukta mRNA özelliği tahmini görevlerinin çoğunda önceki modellerden daha iyi performans göstererek en son teknolojiyi göstermektedir. Ayrıca, çeşitli ilgili görevlerde büyük protein modellerini de geride bırakmaktadır. Sonuç olarak, mRNABERT’in bu çeşitli görevlerdeki üstün performansı, mRNA araştırmaları ve terapötik geliştirme alanında önemli bir sıçramayı göstermektedir.
Son yıllarda, mRNA tedavileri gen terapisinde önemli bir potansiyele sahip devrim niteliğinde bir teknoloji olarak ortaya çıkmıştır 1. Zika virüsü 3, insan immün yetmezlik virüsü 4, influenza virüsü 5, sitomegalovirüs 6, solunum sinsityal virüsü 7, suçiçeği-zona virüsü 8 ve kuduz virüsü 9 dahil olmak üzere çok çeşitli virüslerle mücadele etmek için çok sayıda mRNA aşısı geliştirilmiştir 2. Özellikle , iki COVID-19 mRNA aşısının hızlı bir şekilde geliştirilmesi ve dağıtılması, bu potansiyelin bir kanıtı olarak durmaktadır ve SARS-CoV- 2 , diğer potansiyel patojenler ve tümörlere karşı yeni bir biyoteknolojik platformun ortaya çıkışını işaret etmektedir 10 , 11 , 12 , 13 . mRNA aşıları, bir hastalığın benzersiz özelliklerine uyum sağlayarak belirli antijenleri kodlamak üzere özelleştirilebilir olma konusunda dikkate değer bir yetenek sergilemiştir 2 . DNA aşılarının aksine, mRNA aşıları konak genomu içindeki insersiyonel mutagenezi riskini azaltır 14 ve aynı zamanda hedeflenen antijenin değiştirilebilir ekspresyonunu kolaylaştırır 15 , 16. Endüstriyel bir bakış açısından, in vitro transkripsiyon reaksiyonlarının dikkate değer verimliliği, hücresiz metodolojiler yoluyla mRNA aşılarının hızlı ilerlemesini ve büyük ölçekli üretimini destekler 17 , 18 , 19 ve bu da onları uygun maliyetli bir çözüm haline getirir. Sonuç olarak, mRNA terapötiklerinin hastalıkların önlenmesi ve tedavisindeki uzun vadeli beklentileri giderek daha belirgin hale gelmekte ve tasarımcı ilaçların yeni bir çağını müjdelemektedir 20 .
mRNA, genetik bilgiyi DNA’dan protein sentez makinesine taşımaktan sorumlu özel bir RNA molekülü türüdür. Çekirdeği, proteinleri kodlayan, 5′ ve 3′ çevrilmemiş bölgelerle (UTR’ler) çevrili ve bir 7-metilguanozin (m7G) 5′ başlığı ve bir 3′ poli(A) kuyruğu 21 ( Şekil 1A ) ile stabilize edilmiş bir kodlama dizisinden (CDS) oluşur . Bu bölgelerin yapısal bütünlüğü ve işlevsel sinerjisi, hücreler içinde protein ifadesinin verimli bir şekilde çevrilmesini ve düzenlenmesini sağlar. mRNA bileşenleri yüksek tasarım esnekliği gösterse de, etkili mRNA terapötikleri tasarlamak karmaşık olmaya devam etmektedir 22 , 23 , 24 . Nükleotid dizilerinin, nükleotid modifikasyonlarının ve RNA yapılarının çeviri verimliliğini ve mRNA stabilitesini etkilemek üzere nasıl etkileşime girdiğinin kapsamlı bir şekilde anlaşılması, mRNA tabanlı terapötik protein üretimini optimize etmek için çok önemlidir 20 .

Son zamanlarda, geniş etiketsiz metin üzerinde önceden eğitilmiş ve belirli görevler için ince ayarlanmış büyük dil modelleri (LLM’ler) kullanma konsepti 25 proteinler 26 , 27 , 28 , DNA 29 , 30 ve kodlamayan RNA 31 , 32 , 33 , 34 , 35 , 36 dahil olmak üzere biyolojik dizilere genişletildi . Bu gelişme, mRNA’nın geniş dizisini ve yapısal alanını kapsamlı bir şekilde keşfetmede geleneksel deneysel ve hesaplamalı yöntemlerin sınırlamalarına umut verici bir çözüm sunmaktadır 37 . mRNA molekülleri, deneysel araştırmalarda sıklıkla önemli zorluklar ortaya çıkaran karmaşık analiz teknikleri gerektiren çeşitli mekanizmalar ve etkileşimler sergiler. mRNA ve diğer biyolojik diziler arasındaki nükleotid kompozisyonu ve dizi motiflerindeki benzerlikler göz önüne alındığında, önceden eğitilmiş modeller mRNA araştırmalarını önemli ölçüde geliştirmek için iyi bir konumdadır.
Biyolojik dizileri dil modelleri aracılığıyla çözmeye olan ilgi giderek artmasına rağmen, mRNA dizilerinin semantik gösterimlerini öğrenmek için özel olarak tasarlanmış dil modellerinde kayda değer bir eksiklik devam etmektedir. Mevcut mRNA dil modellerinin pratik uygulaması üç büyük zorlukla karşı karşıyadır. İlk olarak, kamuya açık mRNA dizi verileri nispeten sınırlıdır ve kalite açısından önemli değişkenlik göstermektedir. Bu eksiklik, hem ön eğitim hem de ince ayar aşamalarında model performansını artırmak için hayati önem taşıyan kapsamlı mRNA kütüphanelerinin oluşturulmasını baltalamaktadır. Şu anda, RNA temel modelleri, RNA alanı içinde benzersiz ve ayrı bir varlık olan mRNA’yı hariç tutarak, ön eğitim için öncelikle RNAcentral 38 veya Rfam 39 gibi kaynaklardan gelen büyük ölçekli kodlamayan RNA veri kümelerine dayanmaktadır. İkinci olarak, mRNA dizilerini makine öğrenmesi tekniklerini kullanarak çözmeye yönelik mevcut çabalar, büyük ölçüde her mRNA bölgesini bağımsız bir örnek olarak ele alan belirli UTR 40 , 41 veya CDS 42 , 43 bölgeleri için özel modeller geliştirmeye odaklanmaktadır . Bu modeller, birden fazla bileşenin (5’UTR, CDS, 3’UTR) çeviri sırasında sinerjik olarak işlev görmesi nedeniyle klinik mRNA tasarım zorluklarını ele almak için yetersizdir. Sonuç olarak, hem bölgeler içindeki yerel örüntüleri hem de tüm mRNA dizisini kapsayan küresel örüntüleri etkili bir şekilde yakalayabilen entegre bir tasarım yaklaşımına acil ihtiyaç vardır. Son olarak, model performansını artırmaya yönelik geleneksel yaklaşımlar, model boyutunu 26 , 27 veya eğitim verilerinin hacmini 44 artırmayı içerir ; bu da zaman alıcı ve kaynak yoğun bir süreçtir. Protein mühendisliği, ekleme düzenleme ve gen ifadesi düzenlemesi gibi karmaşık biyolojik süreçlerde, yalnızca mRNA dizi bilgisini değil, aynı zamanda çeşitli biyomoleküller arasındaki etkileşimleri de dahil etmek esastır 45 . Bazı ön hipotezler, nükleotid ve kodon bilgisi 43 , amino asitler ve kodlama dizileri 42 dahil olmak üzere çeşitli biyolojik verilerin entegre edilmesinin mRNA modeli yeteneklerini artırabileceğini öne sürse de , henüz kesin bir yol belirlenmemiştir.
Mevcut RNA ile ilgili dil modelleri, yukarıda belirtilen sorunlar nedeniyle bazı bariz sınırlamalara sahiptir. RNAcentral’dan ncRNA üzerinde önceden eğitilmiş RNABERT 33 , RNAFM 32 , RNAErnie 31 ve ERNIE-RNA 34 , çeşitli ncRNA tahmin görevlerinde yararlılık göstermiştir. MSA tabanlı BERT stili kullanılarak Rfam üzerinde önceden eğitilmiş RNA-MSM 35 , RNAcentral, Rfam ve Nucleotide gibi veritabanlarının bir kombinasyonundan kaynaklanan 36 milyon ncRNA dizisi üzerinde önceden eğitilmiş RiNALMo 36 , hem veri seti boyutu hem de model parametrelerinde ilerlemeleri temsil eder. Hem ncRNA hem de mRNA’yı kapsayan birden fazla kaynaktan çeşitli RNA dizileri üzerinde önceden eğitilmiş UNI-RNA 46 , açık kaynaklı bulunmaması nedeniyle erişilemez durumdadır. mRNA’ya özgü modeller alanında, birden fazla türün 5’UTR dizileri üzerinde önceden eğitilmiş UTR-LM 40 , çeviri verimliliği ve mRNA ekspresyon seviyeleri gibi 5’UTR ile ilgili görevlerde üstündür. Ancak, sınırlı model boyutu ve ön eğitim veri seti önyargısı nedeniyle diğer mRNA alt akış görevlerindeki performansı değerlendirilmeyi beklemektedir. Protein kodlayan DNA (cDNA) üzerinde eğitilmiş CaLM 42 , kodonlar aracılığıyla zengin dizi bilgisini yakalar ve protein mühendisliği testlerinde olağanüstü performans gösterir. Benzer şekilde, girdi olarak kodonlar kullanılarak birden fazla türden CDS dizileri üzerinde eğitilmiş CodonBERT 43 , çeşitli CDS tahmin görevlerinde ustadır. Ancak, her iki model de kodon tabanlı bir belirteçleyici kullanır; bu, bölgeler üçün katları değilse tam uzunluktaki mRNA kodlanırken uygunsuz segmentasyona ve bilgi karışıklığına yol açabilir. Bu, tek nükleotid çözünürlüğünün kaybına yol açarak UTR ile ilgili görevler için değerli bilgilerin çıkarılmasını zorlaştırır. İnsan 3’UTR dizileri üzerinde k-mer belirteçleyici kullanılarak önceden eğitilen 3UTRBERT 41 , belirli 3’UTR görevlerinde diğer yöntemlerden daha iyi performans gösterir. Ancak, çoğu mRNA görevine uygulanabilirliği, 512’lik maksimum giriş uzunluğuyla sınırlıdır (daha fazla ayrıntı için Ek Bilgiler Bölüm 1’e bakın. İlgili Çalışma). Ayrıca, mevcut yoğun Transformatör mimarisi, model genişliği 47 olan ikinci dereceden ölçeklemesi nedeniyle giriş dizisi uzunluğu arttıkça yüksek hesaplama maliyetleriyle karşı karşıyadır . Bu nedenle, model mimarisini ayarlamak ve mRNA ön eğitim modellerinin yeteneklerini artırmak için yenilikçi stratejiler kullanmak gerekmektedir.
Bu gözlemler ışığında, 18 milyondan fazla yedekli olmayan mRNA dizisinden oluşan çeşitli ve yüksek kaliteli bir veri kümesi üzerinde önceden eğitilmiş sağlam bir dil modeli olan mRNABERT’i geliştirdik (yazarlar tarafından düzenlenmiş, daha fazla ayrıntı Yöntemler bölümünde verilmiştir). Önceki modellerin sınırlamalarının üstesinden gelmek için birkaç gelişmiş teknik kullandık. İyi kurulmuş BERT mimarisi 25 üzerine inşa edilen mRNABERT, uzun giriş dizilerini işlemek için geleneksel konumsal gömmeleri Doğrusal Önyargılı Dikkat (ALiBi) 48 ile değiştirdi ve hesaplama verimliliğini artırmak için Flaş Dikkat 49’u entegre etti . Dahası, mRNABERT, bireysel nükleotidleri UTR’ler için belirteçler ve kodlama dizileri için kodonlar (CDS) olarak ele alan yenilikçi bir çift belirteçleme stratejisine sahipti. Bu benzersiz belirteçleme yaklaşımı, yalnızca mRNA’nın biyolojik özellikleriyle uyumlu olmakla kalmıyor, aynı zamanda çok çeşitli alt akış görevleri için de güçlü bir temel oluşturuyor (Şekil 1A ). Ek olarak, mRNA ve protein dizilerini gizli uzayda hizalamak için özelleştirilmiş bir karşılaştırmalı öğrenme şeması sunduk (Şekil 1B ), bu da mRNABERT’in protein fonksiyonları ve mRNA-protein etkileşimleri tahminlerini iyileştirmesine olanak sağladı. Genetik diziler ve protein dizileri arasındaki karmaşık ilişkileri etkili bir şekilde yakalayan bu yöntem, biyolojik süreçlere ilişkin anlayışımızı geliştiriyor ve modelin uygulama yelpazesini genişletiyor.
Sonuçlar
mRNABERT ve kıyaslamalara genel bakış
Tam uzunluktaki mRNA dizilerinin geniş ve entegre veri kümesinden tam olarak yararlanmak için mRNABERT, tüm dizileri kodlamak üzere yeni bir ikili belirteçleme şeması sunar. Dil modellemesinde kritik bir ilk adım olan belirteçleme, modelin yakalayabileceği anlamsal bilgi türlerini belirler. RNA dizileri dört nükleotit bazından oluşur ve geleneksel dil modelleri genellikle, bir modelin bir dizi içindeki nükleotit etkileşimlerini anlamasına yardımcı olabilecek dikkat ağırlıklarını öğrenmek için her nükleotidi bağımsız bir belirteç olarak kodlayan karakter tabanlı belirteçleyiciler kullanırdı. Ancak, tam uzunluktaki mRNA dizileri kodlanırken, belirteç girdisindeki maksimum uzunluk kısıtlamaları modelin temsil kapasitesini tehlikeye atar. mRNA kodonlarının doğası göz önüne alındığında, CDS tabanlı modeller genellikle her kodonu bağımsız bir belirteç olarak ele alır ve bu da bireysel nükleotit bilgilerinin tamamen kaybolmasına neden olur. Sonuç olarak, bu modeller yalnızca belirli dizileri işlemek için uygundur. Bu sorunu çözmek için, mRNA’nın her bölgesini segmentlere ayırmak, yerel özellikleri birden fazla ayrıntı düzeyinde iyileştirmek ve tüm dizinin genel özelliklerini entegre etmek için uygun bir yöntem kullanıyoruz. Bu yaklaşım, çok çeşitli alt akış görevleri için uygun, kapsamlı bir yerleştirme sağlar.
Model mimarisi açısından, mRNABERT, Transformers tabanlı 12 çift yönlü kodlayıcı bloğu üzerine kurulmuştur. Mevcut modellerin giriş uzunluğu sınırlamalarının üstesinden gelmek için, konumsal bilgileri kodlamak için alternatif bir yöntem olan ALiBi’yi tanıttık. Doğrusal sapmaları doğrudan dikkat puanlarına dahil ederek, ALiBi modelin uzun dizileri işleme yeteneğini ve genel performansını artırır. Ayrıca, hassas standart dikkat hesaplamalarını daha zaman ve bellek açısından verimli bir şekilde uygulamak için G/Ç-bilinçli Flash dikkati kullandık ve böylece mRNABERT’in eğitim sürecini hızlandırdık.
mRNA ve protein dizileri arasındaki sıkı işlevsel etkileşimi göz önünde bulundurarak, BERT eğitiminin temel bir parçası olan maskeli dil modeli (MLM) öğrenme aşamasını takiben kodon ve amino asit dizilerini hizalamak için karşılaştırmalı öğrenmeyi dahil ederek yaklaşımımızı geliştirdik. Bu adım, modelin karmaşık biyolojik manzaraya ilişkin anlayışını zenginleştirmeyi amaçlamıştır. Özellikle, mRNABERT’in performansı, karşılaştırmalı öğrenmenin uygulanmasının ardından belirgin bir iyileşme göstermiştir. Model mimarisi ve eğitim süreci hakkında daha fazla bilgi Yöntemler bölümünde verilmektedir.
Çeşitli görevlerde mRNABERT’in önde gelen modellere karşı karşılaştırmalı bir analizini gerçekleştirdik. Sekiz 5’UTR ribozom yükü tahmin görevi için, sinir ağı temel hatları Optimus 50 , FramePool 51 ve MTtrans 52’nin yanı sıra en son teknoloji UTR-LM 40’ı içerir . Altı CDS ile ilgili tahmin görevinde, mRNABERT Codon2vec 53 , TextCNN 54 ve önceden eğitilmiş CDS modeli CodonBERT 43 gibi yöntemlerle değerlendirildi. 3’UTR görevleri için, CNN tabanlı iDeepE 55 ve DeepCLIP 56 ve dil modeli BERT-RBP 57 dahil olmak üzere üç temel yöntem kullanarak 22 RBP bağlanma bölgesini tahmin etmeye odaklandık . m 6 A bölge tahmin görevinde, dokuz farklı hücre hattından veri topladık ve mRNABERT’i SRAMP 58 ve WHISTLE 59 gibi makine öğrenme yöntemleriyle karşılaştırdık . Her iki görev de özel olarak tasarlanmış 3UTRBERT 41’i içeriyor . Ayrıca, yukarıda belirtilen tüm ncRNA modellerinin bu görevlerdeki performansını değerlendiriyoruz. Proteinle ilgili görevler, erime noktası ve çözünürlük tahmininin yanı sıra yedi tür boyunca transkript bolluğu tahminini içerir. Burada, mRNABERT, ESM2 27 , ProtTrans 26 , Ankh 28 ve önceden eğitilmiş kodon dili modeli CaLM 42 dahil olmak üzere yüksek performanslı protein modelleriyle kıyaslandı . Son olarak, sekiz tam uzunlukta mRNA özelliği tahmin görevi için tüm mevcut RNA ile ilgili önceden eğitilmiş modelleri kıyasladık. Bu kapsamlı karşılaştırma, mRNABERT’in tüm görevlerdeki olağanüstü performansını göstermektedir.
mRNA’nın çok boyutlu biyolojik bilgilerinin yakalanması
mRNABERT’in dizilerden çoğu temel modelden daha fazla biyolojik bilgi öğrenebildiğini göstermek için, onun gömülü yapılarının bir analizini gerçekleştirdik ve biyolojik dizilerden işlevsel ve evrimsel bilgiyi nasıl çıkardığını karakterize ettik.
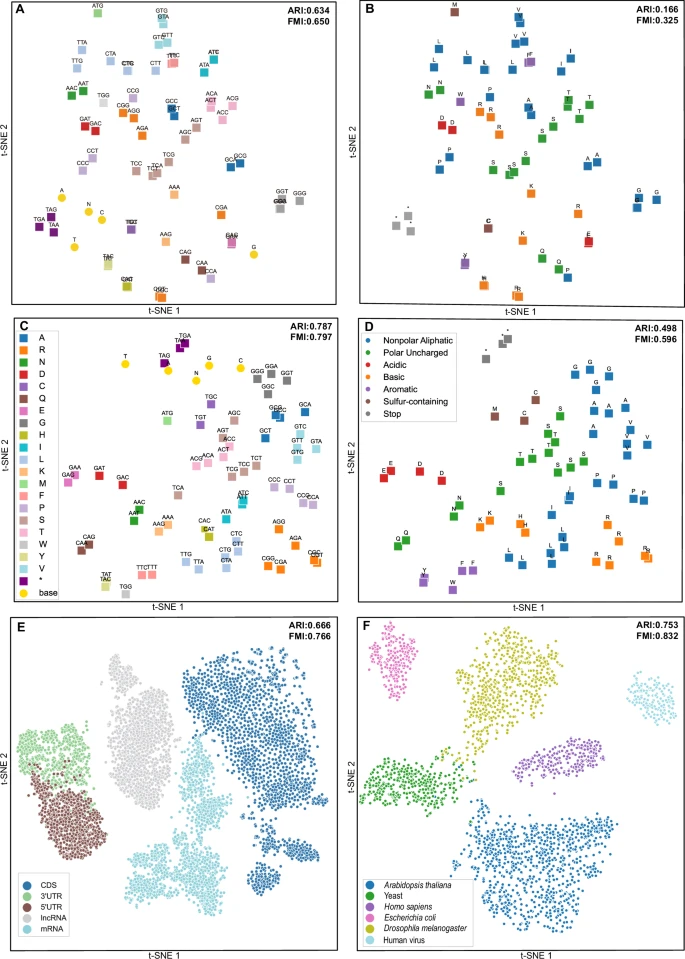
Araştırdığımız ilk husus, modelin kelime dağarcığı temsil yetenekleriydi ve genetik kodun temel biyolojik prensiplerini ayırt etme yeteneğine odaklandı. İdeal olarak, bir mRNA modeli eşanlamlı kodonlar arasındaki benzerlikleri belirlemelidir, ancak bu görev, açıklama yapılmamış ön eğitim verileri ve modelin biyolojik dizileri, nükleotidler veya kodonlar hakkında açık bilgi içermeyen belirteçler olarak temsil etmesi nedeniyle zordur. Ek olarak, karşılaştırmalı öğrenmenin etkinliğini doğrulamak için ablasyon deneyleri yürüttük. Şekil 2 A, B’de gösterildiği gibi , karşılaştırmalı öğrenmesi olmayan mRNABERT modeli amino asit düzeyinde düzensiz kümeleme sergiledi. Ancak, mRNABERT’in kelime dağarcığı gömmelerine aynı t-SNE 60 boyut indirgemesi ve bunları iki boyutlu bir uzaya yansıtma yoluyla, aynı amino asit tipine karşılık gelen eşanlamlı kodonların birlikte kümelenme eğiliminde olduğunu gözlemledik (Şekil 2C ). Bu kümeleme, modelin eğitildiği kapsamlı verilerden genetik kodu başarıyla öğrendiğini göstermektedir. Ayrıca, amino asitleri farklı kimyasal özelliklerine göre ayırt etmek için renk kullanarak (Şekil 2D ), modelin benzer özelliklere sahip amino asitleri etkili bir şekilde gruplandırdığını, ARI’nin 0,166’dan 0,498’e ve FMI’nin 0,325’ten 0,596’ya çıktığını bulduk (Yöntemler). Bu, karşılaştırmalı öğrenmenin modelin amino asitler hakkında ek anlamsal bilgi yakalamasını sağladığını açıkça göstermektedir.
Daha sonra, mRNABERT’in çeşitli RNA verisi türlerini sınıflandırma yeteneğini değerlendirdik. Şekil 2E’de gösterildiği gibi , mRNABERT, 5’UTR ve 3’UTR dizileri de dahil olmak üzere farklı mRNA bölgeleri arasında başarıyla ayrım yaptı. Etkileyici bir şekilde, ncRNA verileri üzerinde açıkça eğitilmemiş olmasına rağmen, uzun kodlamayan RNA (lncRNA) ve mRNA dizileri arasında ayrım yapma yeteneği de gösterdi. Bu, mRNABERT’in yeterli biyolojik özellikleri kapsülleme kapasitesini vurgulayarak, yalnızca çeşitli mRNA bölgeleri arasında ayrım yapmasını değil, aynı zamanda mRNA’yı diğer RNA dizisi türlerinden de ayırt etmesini sağlar. Tüm mRNA dizisinden derinlemesine anlamsal bilgi çıkararak, salt uzunluğun ötesine uzanan dizi benzerliklerini belirler.
Daha sonra analizimiz, çeşitli veri kümelerindeki geniş bir biyolojik sınıflandırma yelpazesini temsil edecek şekilde özenle seçilmiş altı farklı türden türetilen dizilerin gömülmelerine odaklandı. Şekil 2F’de gösterilen saçılma grafiği , farklı türleri birbirinden ayıran net sınırlarla, homolog dizilerin net bir kümelenmesini ortaya koymaktadır. Bu sonuç, mRNABERT’in biyolojik dizilere gömülü evrimsel bilgileri tanıma ve saklama yeteneğini vurgulayarak, dizi düzeyinde biyolojik ayrıntıları yakalama konusundaki güçlü yeteneğini vurgulamaktadır.
5′UTR dizilerinden ribozom yükünün tahmini
Çeviri verimliliğinin kontrolü, 5′UTR dizisinin kritik rolüne bağlıdır. Herhangi bir anda bir mRNA molekülüne bağlı ribozom sayısı olarak tanımlanan ribozom yükü, protein sentezi verimliliğinin temel bir belirtecidir 50 . Bu nedenle, 5′UTR dizilerinden ribozom yükünü doğru bir şekilde tahmin etmek, özellikle mevcut 5′UTR şablonlarının ötesinde yeni diziler oluştururken, protein ekspresyonunu en üst düzeye çıkarmak için mRNA dizi tasarımını optimize etmek açısından son derece önemlidir.
Bu zorluğun üstesinden gelmek için, ilgili ribozom yükleriyle birlikte 280.000 gen dizisi içeren bir kütüphane oluşturmak üzere büyük ölçekli paralel muhabir analizleri (MPRA) kullanan önceki çalışmalardan alınan bir kıyaslama veri kümesinden yararlandık 50 . Yaklaşımımız, 5’UTR dizilerinden ribozom yükünü tahmin etmek için mRNABERT modelini ince ayarlamayı içeriyordu (“Yöntem” bölümünde ayrıntılı olarak açıklanmıştır). mRNABERT’in yanı sıra, Optimus 50 , FramePool 51 ve MTtrans 52 dahil olmak üzere bu görev için özel olarak tasarlanmış çeşitli makine öğrenme modellerini ve ayrıca UTR-LM 40 , RNABERT 33 ve RNA-FM 32 gibi önceden eğitilmiş dil modellerini kıyasladık. mRNABERT’in performansı, sekiz sentetik kütüphanedeki kıyaslama yöntemleriyle karşılaştırılarak değerlendirildi.
Çalışmamızın sonuçları, Şekil 3’te ve Ek Tablolar 4 ve 5’te gösterildiği gibi , en iyi performansı gösteren uzmanlaşmış model olan UTR-LM ile karşılaştırılabilir olan mRNABERT’in olağanüstü performansını vurgulamaktadır. Özellikle, en büyük MPRA veri kümelerinde ( U 1 ve U 2 olarak gösterilen sabit uzunluktaki rastgele UTR’ler ), modelimiz en son teknoloji sonuçlarına ulaşmıştır (Spearman R = 0,962 ve 0,924). Kalan altı veri kümesinde, modelimiz üç görevde ( Ψ 2 , m1 Ψ 1 ve mC-U 1 ) liderlik etmiş ve en iyi performansa sahip görev sayısı bakımından UTR-LM ile etkili bir şekilde eşleşmiştir (her ikisi de 8’den 4’ünde en iyi sonuçları elde etmiştir).

CDS tahmin görevlerinde mRNABERT’in değerlendirilmesi
Modelimizin CDS tahmin görevlerindeki performansını değerlendirmek için birden fazla veri kümesi topladık. Bu veri kümeleri, mantarlar ve E. coli’deki protein ekspresyonuna ilişkin binlerce veri noktasından oluşan mRFP Ekspresyonu 61 , Mantar Ekspresyonu 62 ve Escherichia coli Proteinleri 63 veri kümelerini ; mRNA stabilite verilerini içeren mRNA Stabilitesi 64 ve SARS-CoV-2 Aşı Bozulması 65 veri kümelerini; ve tetrasiklin riboswitch dimer dizilerini vurgulayan Tc-Riboswitch’ler 66 veri kümesini içerir. Bu veri kümeleri, yeni yayınlanan rekombinant proteinlerden SARS-CoV-2 aşı tasarımı için biyo-hesaplamaya kadar uzanan verileri içeren, mRNA translasyonu, stabilitesi ve düzenlenmesiyle ilgili çeşitli alt akış görevlerini kapsar (Ek Tablo 6 , veri kümeleri hakkında ayrıntılı bilgi içerir).
Bu veri kümelerinde mRNABERT’i ince ayarladıktan sonra, performansını TF-IDF 67 , TextCNN 54 , Codon2vec 53 , RNABERT 33 , RNA-FM 32 ve CodonBERT 43 dahil olmak üzere çeşitli son teknoloji CDS tahmin yöntemleriyle karşılaştırdık . Sonuçlarımız, mRNABERT’in tüm 6 CDS ile ilgili tahmin görevinde diğer tüm yöntemleri geride bıraktığını veya onlarla eşleştiğini, SARS-CoV-2 Aşı Degradasyonu veri kümesinde olağanüstü bir performans gösterdiğini göstermektedir (Tablo 1 ).Tablo 1 Spearman korelasyonunu kullanarak CDS alt akış görevlerinde performans karşılaştırması
Ayrıca, analizimiz CodonBERT gibi kodon tabanlı modellerin protein ekspresyonu görevlerinde mükemmel olduğunu ancak stabilite ile ilgili görevlerde vasat bir performans sergilediğini ortaya koydu. Bu tutarsızlık, kodonların protein ekspresyonunda oynadığı önemli role atfedilebilir 61 , oysa mRNA stabilitesi ikincil yapısıyla yakından bağlantılıdır 64 . Özellikle, kodon tabanlı modellerin performansı, RNA dizilerinin yerel ve küresel ikincil yapı örüntülerinin kritik olduğu veri kümelerinde düştü 68 , örneğin SARS-CoV-2 aşısının bozunması ve Tc-riboswitch veri kümelerinde. Bunun aksine, mRNABERT nükleotid ve kodon bilgilerini etkili bir şekilde entegre ederek yapısal olarak ilgili 5’UTR ve 3’UTR bölgelerini kodlar. Sonuç olarak, CodonBERT’in zorlandığı görevlerde üstün performans gösterir, çünkü milyonlarca mRNA dizisinden ortak evrimsel ve yapısal özellikleri öğrenebilir. Bu yetenek, oldukça ifade edici ve stabil mRNA dizilerinin tasarlanmasına yardımcı olur.
3′UTR dizilerinden RBP bağlanma bölgelerinin tespiti
RNA bağlayıcı proteinler (RBP’ler) spesifik olarak RNA moleküllerine bağlanır ve bu bağlanma hem RNA dizilerine hem de mekansal yapı özelliklerine bağlıdır 69 . 22 RBP için protein-RNA çapraz bağlama bölgelerini indirip işledik 70 ve bu deneysel olarak belirlenen verileri kullanarak RBP bağlanma bölgelerini tahmin etmek için mRNABERT’i ince ayarladık. Modelimizin öngörü performansını değerlendirirken, onu iDeepE 55 , DeepCLIP 56 , RPI-Net 71 , GraphProt2 72 , BERT-RBP 57 sinir ağı modelleri, RNABERT 33 ve RNAFM 32 gibi önceden eğitilmiş tüm RNA modelleri ve daha önce 3’UTR görevleri için tasarlanmış en iyi model olan 3UTRBERT 41 dahil olmak üzere çeşitli hesaplama yöntemleriyle kıyasladık .
Her modelin etkinliğini değerlendirmek için beş katlı çapraz doğrulama kullandık ve tahminleri üç ölçüt kullanarak değerlendirdik: doğruluk (ACC), F1 puanı ve Matthews korelasyon katsayısı (MCC) (Değerlendirme ölçütlerinin tanımı Ek Tablo 7’dedir ) . 22 RBP’nin tamamında, mRNABERT ortalama 0,786 ACC, 0,751 F1 puanı ve 0,501 MCC ile üstün performans gösterdi; bu, ortalama 0,785 ACC, 0,751 F1 puanı ve 0,503 MCC ile en iyi uzmanlaşmış 3UTRBERT modeliyle karşılaştırılabilir. Dikkat çekici bir şekilde, mRNABERT 22 RBP’nin 13’ünde diğer yöntemlerden daha iyi performans gösterdi ve 9 RBP için 3UTRBERT’in performansını aştı. 3UTRBERT hariç, mRNABERT diğer tüm modellerden önemli ölçüde daha iyi performans gösterdi. Bir sonraki en iyi performans, 0,758’lik ACC, 0,565’lik F1 puanı ve 0,413’lük MCC ile iDeepE tarafından elde edildi; bu değerler, mRNABERT’inkinden ortalama %20 daha düşüktü (Şekil 4A ve Ek Tablo 8 ). BERT-RBP’nin ön eğitim eksikliğinden dolayı geride kaldığını, diğer derin öğrenme yöntemlerinin ise yetersiz model kapasitesi nedeniyle düşük performans gösterdiğini belirtmekte fayda var. Bu karşılaştırmalı sonuçlar, mRNABERT’in 3′UTR’deki RBP bağlanma bölgelerini doğru bir şekilde belirlemek için oldukça etkili bir yöntem olduğunu göstermektedir.

3′UTR dizilerinden m 6 A modifikasyon bölgelerinin belirlenmesi
N6-metiladenozin (m 6 A), hücrelerdeki en yaygın kovalent modifikasyondur ve çok sayıda kritik gelişimsel süreçte ve insan hastalıklarında rol oynar 73 . m 6 A-Atlas veritabanından gerçek m 6 A modifikasyon bölgelerini indirdik 74 ve ince ayar yaparak mRNABERT’in potansiyel m 6 A modifikasyon bölgeleri için tahmin yeteneklerini geliştirdik (ayrıntılı bilgi için Yöntemler bölümüne bakın).
mRNABERT’in öngörü performansının literatürde bulunan çeşitli modellerle, örneğin en etkili model olan 3UTRBERT 41 ve farklı makine öğrenimi tabanlı yöntemler (SRAMP 58 , WHISTLE 59 , iMRM 75 ) ve derin öğrenme tabanlı yöntemlerle (DeepM6ASeq 76 ) karşılaştırmalı bir analizini gerçekleştirdik. Şekil 4B ve Ek Tablo 9’da gösterilen sonuçlar , mRNABERT’in tüm dokuz hücre hattında tutarlı bir şekilde ikinci en iyi performansı elde ettiğini, önde gelen 3UTRBERT modelinin hemen arkasında yer aldığını ve diğer tüm modelleri geride bıraktığını gösterdi. Bu bulgular, mRNABERT’in 3’UTR’den yapısal ve işlevsel bilgileri yakalama ve kullanma yeteneğine sahip olduğunu ve yalnızca 3’UTR verileri üzerinde önceden eğitilmiş modellerle karşılaştırılabilir bir performans sergilediğini göstermektedir.
Ekleme yerlerinin ve alternatif poliadenilasyonun tahmini
RNA ekleme, ökaryotik gen ifadesinde temel bir düzenleyici mekanizmadır ve öncül mRNA’lardan (pre-mRNA’lar) kodlamayan intronik dizilerin hassas bir şekilde çıkarılmasını ve olgun transkriptler oluşturmak için kodlayıcı ekzonların bağlanmasını düzenler 77 . Bu süreç, ekzon-intron sınırlarını belirleyen ekleme bölgelerinin doğru bir şekilde tanınmasına kritik derecede bağlıdır. İntronların 5′ ucundaki verici bölgeler eklemeyi başlatırken, 3′ ucundaki alıcı bölgeler ekzon bağlanmasını kolaylaştırır.
Bu ekleme bölgelerinin doğru bir şekilde tanımlanması, gen mimarisi ve transkripsiyonel izoformların belirlenmesi için kritik bir ön koşul oluşturur. Bu zorluğa yönelik hesaplamalı yaklaşımlar sıklıkla, algoritmik modellerin pre-mRNA molekülleri içindeki sahte dizilerden gerçek ekleme sinyallerini ayırt ettiği dizi tabanlı ikili sınıflandırma görevleri olarak çerçevelenir. Bu amaçla, dört farklı türden donör ve alıcı bölge verilerini içeren, yaygın olarak kabul gören pozitif ve negatif ekleme bölgesi dizilerinden oluşan bir veri setini kullandık 78 . Tüm RNA temel modellerini değerlendirmek için aynı veri setini ve test protokolünü kullanarak modelleri ince ayarladık. mRNABERT, yalnızca ERNIE-RNA tarafından geride bırakılan ve hem RiNALMo hem de UNI-RNA’yı geçen ikinci en yüksek genel performansı sergiledi (Ek Tablo 10 ).
Alternatif poliadenilasyon (APA), seçici 3′UTR işleme yoluyla transkriptomları çeşitlendiren yaygın bir transkripsiyon sonrası düzenleyici mekanizmadır 79 ve böylece farklı stabilite, lokalizasyon ve protein kodlama potansiyeline sahip mRNA izoformları üretir. Bu dinamik süreç, gen ekspresyon ağlarını ince ayarlar ve hücresel farklılaşma, stres tepkileri ve gelişimsel örüntüleme için vazgeçilmezdir.
APA dinamiklerini sistematik olarak ölçmek için, BEACON veri seti 80’den türetilen izoform düzeyindeki tahminleri analitik çerçevemize entegre ettik. Yaklaşımımız, özellikle açıklamalı 3′UTR bölgelerindeki proksimal ve distal poliadenilasyon bölgelerinin (PAS) göreceli kullanımını modelleyerek, APA aracılı düzenleyici sonuçların hassas bir şekilde çözümlenmesini sağlar. Bu görevde, mRNABERT diğer tüm RNA temel modellerine göre önemli bir avantaj sergilemiştir (Ek Tablo 11 ).
mRNABERT’in bu özel görevlerdeki üstün performansı, transkripsiyon sonrası mRNA modifikasyonlarına ilişkin derin anlayışına dair ikna edici kanıtlar sunmakta ve böylece mRNA araştırmalarının daha geniş manzarası içinde analitik yeteneklerini önemli ölçüde genişletmektedir.
mRNABERT’in protein mühendisliği görevlerine uygulanması
mRNABERT’in proteinle ilgili görevlerdeki performansını değerlendirdik ve kodon pLM modellerinin daha önce belirli amino asit dizisi açıklama görevlerinde üstün sonuçlar gösterdiğini belirttik 42 . mRNABERT’in performansını, özellikle protein erime noktalarını ve çözünürlüğünü tahmin etmek gibi çeşitli proteinle ilgili görevlerde değerlendirdik. Ayrıca, modelin temel kodon kullanım görevlerindeki etkinliğini değerlendirmek için yedi organizmadan transkriptom bolluğu verileri topladık ve derledik. Tüm veri kümeleri, Yöntemler bölümünde daha fazla ayrıntı sağlanarak orijinal kodon dizilerine eşlendi.
ESM2 27 , ProtTrans 26 ve Ankh 28 serisi gibi gelişmiş pLM’lere amino asit dizileri besledik ve kodon dizilerini CaLM 42 ve mRNABERT dahil olmak üzere mRNA modellerine eşledik. Ayrıca, amino asit semantik bütünleşmesinin etkisini daha iyi anlamak için mRNABERT’i karşılaştırmalı öğrenme olmadan da test ettik. Bu modellerden elde edilen yerleştirmeler daha sonra alt akış görev modelinde kullanıldı ve performans, beş katlı çapraz doğrulama kullanılarak değerlendirildi (ayrıntılı bilgi için Yöntemler bölümüne bakın).
Şekil 5 ve ek dosya, karşılaştırmalı öğrenme özelliğine sahip mRNABERT’in tüm görevlerde önemli gelişmeler gösterdiğini göstermektedir. Erime noktası tahmininde (Şekil 5A ), karşılaştırmalı öğrenmeden sonra mRNABERT, R2 değerini 0,60’tan 0,77’ye çıkardı; bu, CaLM’nin 0,78’inin biraz altında kalsa da diğer tüm büyük ölçekli protein modellerini (R2 = 0,73 olan en iyi ProtT5-XL) geride bıraktı . Çözünürlük tahmininde (Şekil 5B ), mRNABERT, 0,63’lük bir R2 değerine ulaşarak hem karşılaştırmalı öğrenmeden önceki performansını hem de CaLM’nin 0,61’ini geride bıraktı. Dahası, mRNABERT’in bu görevdeki performansı çoğu protein modeliyle karşılaştırılabilir olup, ProtT5-XL ve Ankh-large ( R2 = 0,66 ) gibi büyük ölçekli modellerin hemen gerisinde kalmaktadır .

Ek olarak, yedi türde transkript bolluğu tahmini görevinde mRNABERT, beş türde ( E. coli ve Haloferax hvolcanii hariç) CaLM’yi geride bıraktı. Dikkat çekici bir şekilde, birden fazla türde mRNABERT diğer tüm modellerden daha iyi performans gösterdi. Örneğin, Homo sapiens için tahminlerde mRNABERT, 0,38’lik bir R 2 değerine ulaştı ; bu, CaLM’nin 0,35’inden önemli ölçüde daha yüksek ve diğer tüm protein modellerini geride bıraktı (en yüksek R 2 = 0,36). Pichia pastoris ve Saccharomyces cerevisiae tahminlerinde mRNABERT, diğer modellerden önemli ölçüde üstündür ve sırasıyla 0,56 ve 0,53’lük en iyi R 2 değerlerine ulaşırken, protein modelleri arasındaki en yüksek performans 0,53 ve 0,52’dir (Şekil 5C ).
CaLM modelinin başarısı, kodon tabanlı ön eğitimin protein modellerinin kalitesini artırma potansiyelini vurgulamaktadır 42 . Ayrıca, mRNA modelimiz belirli proteinle ilgili görevlerde üstün performans sergilemiştir. Bu sonuçlara ve ablasyon çalışmalarına dayanarak, amino asit bilgilerinin kodlama dizileriyle bütünleştirilmesi, genel model performansını önemli ölçüde artırmak için uygun maliyetli bir yaklaşım olarak ortaya çıkmaktadır. Bu sonuç, makine öğrenimi yeteneklerini geliştirmek için kapsamlı biyolojik verilerden yararlanmanın, böylece model sınırlamalarını ele almanın ve uygulanabilirliğini genişletmenin potansiyelini vurgulamaktadır.
Tam uzunluktaki mRNA dizilerini kullanarak uygulanabilirliğin değerlendirilmesi
Tam mRNA dizilerinin stabilitesini ve ekspresyonunu en üst düzeye çıkarmak için yeniden tasarlanması, terapötik mRNA’nın genel performansını önemli ölçüde iyileştirebilir 23 . Ancak, bu tür dizilerin tasarlanması, mRNA dizilerinin ve yapılarının çözelti ve hücrelerdeki ekspresyon ve stabilitelerini nasıl etkilediğine dair sınırlı anlayış nedeniyle zorluklarla karşı karşıyadır 81 , 82 , 83 . Bu nedenle, tam mRNA’nın yapısal ve işlevsel özelliklerini doğru bir şekilde tahmin etmek, mRNA tasarım kurallarının anlaşılmasına yardımcı olacak ve mRNA aşısı geliştirmeyi büyük ölçüde ilerletecektir.
Farklı UTR’ler ve CDS’lere sahip büyük miktarlarda tam uzunlukta mRNA’yı hızla sentezlemek zordur ve bu da bunların kararlılığı ve ifade kabiliyetlerinin yüksek verimli deneysel yaklaşımlarla doğrudan karşılaştırılmasını imkansız hale getirir. Bu sorunu ele almak için, geniş bir UTR ve CDS mRNA dizisi yelpazesini kapsayan yüzlerce haberci gen yapısından oluşan bir veri seti derledik 23 ve 112 ayrı 5′ ve/veya 3′ UTR ve 121 CDS’ye sahip 233 kullanılabilir mRNA dizisiyle sonuçlandı. Veri seti, dört hücre içi çeviri verimliliği ve protein ifade seviyelerini doğrudan etkileyen iki kararlılıkla ilgili özellik için etiketler içeriyordu. mRNA modellerinin potansiyelini daha fazla keşfetmek için, toplanan verileri kullanarak mRNABERT’i ince ayarladık ve gerçek dünya mRNA görevlerindeki performansını değerlendirdik. Ek olarak, 5’UTR’ler için UTR-LM 40 , kodonla ilişkili CaLM 42 ve mRNA-FM, 3’UTR’ler için 3UTRBERT 41 ve RNABERT 33 , RNA-FM 32, RNA-MSM 35 , ERNIE -RNA 34 , RNAErnie 31 ve RiNALMo dahil olmak üzere çeşitli önceden eğitilmiş RNA modelleri gibi mevcut tüm RNA temel modellerini değerlendirdik .
Şekil 6 ve Ek Tablo 12’deki sonuçlar , mRNABERT’in tüm görevlerde diğer modellerden önemli ölçüde daha iyi performans gösterdiğini göstermiştir. ncRNA verileri üzerinde önceden eğitilmiş modeller, tam uzunluktaki mRNA’ya genelleme yapmakta zorlanmış ve belirli mRNA bölgesi görevlerinde mükemmel olan modeller, tam mRNA görevlerinde zayıf performans göstermiştir. Bu tutarsızlık, muhtemelen önceki modellerin maksimum giriş uzunluklarıyla kısıtlanmış nükleotit tabanlı belirteçleyiciler kullanmasından kaynaklanmaktadır ve bu da tam uzunluktaki mRNA’lar için kesilmeye ve bilgi kaybına neden olmaktadır. Kodon tabanlı belirteçleyiciler genellikle üçlü olmayan bölge segmentlerini yanlış yorumlayarak bilgi karışıklığına yol açmaktadır. Modelimiz, UTR ve CDS bölgeleri için çift belirteçleyici yaklaşımını benimsemiş ve bir BERT mimarisinin giriş dizisi uzunluklarını genişletmesini ve pratik uygulamayı iyileştirmesini sağlayan gelişmiş bir teknik içermektedir. Ayrıca, önceki mRNA modelleri belirli parçalar üzerinde eğitilmiş ve değerlendirilmiş, bu da tam uzunluktaki mRNA görevlerindeki etkinliklerini sınırlamıştır. Buna karşılık, ncRNA modelleri öncelikli olarak RNA yapısı tahminine odaklanır ve bu da genellikle mRNA ile eğitilen modelleri aşmayı zorlaştırır.

mRNABERT’in ultra uzun mRNA dizileri için öngörü yeteneklerini titizlikle değerlendirmek amacıyla, memeli hücrelerinde tam uzunluktaki mRNA’nın çeviri verimliliğini tahmin etmeye odaklanan ek kıyaslama görevleri yürüttük 84 . Analiz, 140’tan fazla insan ve fare hücre tipinde eşleşen RNA-seq verileriyle eşleştirilmiş binlerce ribozom profili deneyinden türetilen kapsamlı bir veri setinden yararlandı. Özellikle, insan veri seti (ortalama uzunluk: 4040 nt), 1024 nt’yi aşan dizilerin %94,9’unu içeriyordu ve bunların %82,2’si kodlama sonrası 1022 belirteci aşıyordu. Fare veri seti (ortalama uzunluk: 3645 nt) karşılaştırılabilir oranlar gösterdi (sırasıyla %94,6 ve %80,8). Bu dizi uzunlukları, mevcut RNA modellerinin (tipik olarak 1024 nt ile sınırlıdır) ve eğitim veri setimizin ( Ek Tablo 13 ) maksimum girdi kapasitelerini önemli ölçüde aştı . Ancak, ALiBi uygulamasının mRNABERT’in 1022 token’dan uzun dizileri işlemesini sağladığını vurgulamak önemlidir. Modelin max_length parametresini aşan diziler, hesaplamalı uygulanabilirliği sağlamak için sistematik olarak kesilmiştir.
mRNABERT’in eğitim uzunluğunu aşan diziler üzerindeki genelleme yeteneklerini değerlendirmek için, mRNABERT’i 1022, 2044 ve 3066 belirteçlik maksimum dizi uzunluklarıyla değerlendirdik. Sonuçlarımız, mRNABERT’in hücre tipleri arasında ortalama 0,66 R² değerine ulaşarak mevcut tüm RNA modellerinden istikrarlı bir şekilde daha iyi performans gösterdiğini ortaya koymaktadır (Tablo 2 ). Bu, maksimum 0,42 R² değerine (aralığı: 0,06-0,42) ulaşan önceki RNA modellerine kıyasla 1,6 ila 10,4 kat arasında değişen önemli bir performans artışını temsil etmektedir . Dahası, artan giriş uzunluğuyla gözlenen performans kazanımları, mRNABERT’in daha uzun dizilere sağlamlık ve uygulanabilirlik gösterdiğini düşündürmektedir. Bu bulgu, model tasarımımızın faydalarını vurgulamaktadır: çift belirteçleme kapsamlı mRNA bilgisinin yakalanmasını kolaylaştırırken, ALiBi mekanizması genişletilmiş dizi uzunluklarına genellemeyi mümkün kılmaktadır. mRNABERT, daha uzun mRNA dizilerinin özelliklerini tahmin etmede uygulanabilirlik ve belirgin bir avantaj göstermektedir.Tablo 2 Ultra uzun mRNA dizileri için çeviri verimliliği tahmininin karşılaştırılması
Genel olarak, mRNABERT’in bu zorlu görevlerdeki başarısı, model tasarım stratejimizin etkinliğini ve gerçek dünya uygulama senaryolarındaki muazzam potansiyelini tam olarak göstermektedir.
Tartışma
Bu çalışmada, mRNA özelliklerini analiz etmek ve tahmin etmek için tasarlanmış temel bir model olan mRNABERT’i geliştirdik. Çeşitli türlerden derlediğimiz 18 milyon mRNA dizisinden oluşan yeni repertuarımız üzerinde önceden eğitilen mRNABERT, tüm mRNA ile ilgili görevleri tek bir modelle evrensel olarak ele almanın temelini oluşturur. Dahası, pLM’den türetilen amino asit semantik bilgilerini karşılaştırmalı öğrenme yoluyla entegre eder. Diğer gözetimsiz büyük dil modellerine benzer şekilde, mRNABERT’in amacı, doğal seçilim tarafından şekillendirilen geniş bir bilgi yelpazesini yakalayarak yüksek ifade ve kararlı yapılara sahip mRNA dizilerinin tasarımını kolaylaştırmaktı. Analizlerimiz, mRNABERT’in çeşitli biyolojik dizilerden gerçekten de çok sayıda önemli bilgi edindiğini göstermektedir.
Daha sonra, mRNABERT’in UTR’ler, CDS ve proteinle ilgili veri kümeleri üzerinde yapılan testler de dahil olmak üzere çeşitli denetimli tahmin görevlerindeki performansını değerlendirdik. Her mRNA bölgesi için en son yöntemlerle yapılan kıyaslama karşılaştırmaları, mRNABERT’in çeşitli mRNA görevleri için genellikle en iyi uzmanlaşmış modellerden daha iyi performans gösterdiğini veya onlarla eşleştiğini göstermektedir. Özellikle, mRNABERT, tam uzunluktaki mRNA’nın özelliklerini tahmin etmede her testte diğer tüm modelleri geride bırakarak geniş uygulanabilirliğini ve pratik potansiyelini ortaya koymuştur.
mRNABERT’in başarısına katkıda bulunan birkaç önemli avantaj bulunmaktadır. Güçlü bir ön eğitim modeli geliştirmek, büyük ölçekli ve yüksek kaliteli verilere dayanır. Modelimizin başarısının temelini oluşturan yüksek kaliteli bir mRNA dizi veritabanı oluşturduk. Bu kapsamlı verilerle eğitildikten sonra, model içsel yapıları ve sözdizimini öğrenerek, minimum ince ayar ile belirli alt görevlere esnek bir şekilde uyum sağlamasını mümkün kılar. Mevcut belirteçleştirme yöntemlerinin hesaplama ve temsil kapasitelerindeki sınırlamalarını belirledik ve tam uzunluktaki mRNA için uygun, yenilikçi bir hibrit modelleme yaklaşımı önerdik. Ayrıca, mevcut model yapılarını geliştirmek ve karşılaştırmalı öğrenme yoluyla genel performansı önemli ölçüde artırmak için doğrusal önyargılı dikkat (ALiBi) ve Flaş Dikkat gibi çeşitli teknikleri entegre ettik. Ayrıca, çok modlu veriler kullanılarak mRNA model yeteneklerinin geliştirilmesine yönelik ön çalışmalar da yapıldı.
mRNABERT’in mRNA özelliklerini doğrudan dizilerden doğru bir şekilde tahmin etme yeteneği, araştırmacıların yeni mRNA mekanizmalarını keşfetmelerine yardımcı olacaktır. Odaklanılan görevler doğası gereği denetlenmiş olsa da, büyük dilli bir model olarak mRNABERT, üretimsel amaçlar için de kullanılabilir. Özellikle, bu modeli, hedef protein (amino asit) dizilerine dayalı olarak mRNA aşı dizilerinin çeşitli bileşenlerini optimize etmek veya genom düzenleme gibi belirli biyolojik işlevleri gerçekleştirmek için belirli mRNA dizilerini seçip tasarlamak için kullanmayı planlıyoruz. Gelecekteki araştırmalar ilerledikçe, mRNA tasarımının ve örneklemesinin mRNABERT aracılığıyla optimize edilmesinin temel araştırmalar, hastalık tedavisi ve yeni tedavilerin geliştirilmesi açısından büyük önem taşıyacağını öngörüyoruz.
mRNABERT ile elde edilen ümit verici sonuçlara rağmen, bunun temel bir adım olduğunu kabul ediyor ve gelecekteki iyileştirmeler için heyecan verici birkaç yol olduğunu biliyoruz. Önemli bir yön, yapısal bilginin açıkça bütünleştirilmesidir. Gelecekteki yinelemeler, tahmin edilen mRNA ikincil yapılarını veya diğer biyofiziksel özellikleri içeren çok modlu mimariler geliştirerek, diziden dolaylı çıkarımın ötesine, fiziksel etkileşimlerin doğrudan modellenmesine geçebilir. Bu, özellikle RBP bağlanması gibi görevler için modelin mekanik yorumlanabilirliğini artıracaktır. Mimari açıdan, son derece uzun transkriptlerdeki Transformatörlerin doğasında bulunan hesaplama kısıtlamalarını ele almak için, doğrusal karmaşıklığa sahip modelleri keşfetmek, ölçeklenebilirliği artırmak için ümit verici bir yoldur. Gelecekteki çalışmalar, uzun menzilli bağımlılıkları geleneksel Transformatörlerden daha verimli bir şekilde ele alma konusunda büyük potansiyel gösteren Durum Uzayı Modelleri gibi seyrek dikkat mekanizmalarını veya yeni ortaya çıkan mimarileri araştırabilir. Son olarak, gelecekteki çalışmalar, karmaşık genomik özelliklerin daha ayrıntılı bir şekilde işlenmesini sağlamak için veri ön işleme stratejilerini iyileştirerek büyük ölçekli veri setimize de dayanabilir. Bu yönlerin izlenmesi, gelecek nesil daha güçlü ve kapsamlı mRNA dil modellerinin geliştirilmesi açısından kritik öneme sahip olacaktır.
Sonuç olarak, mRNABERT, biyolojik bilimler ile mevcut bilgi sistemleri arasında köprü kuran, mRNA için öncü bir önceden eğitilmiş modeldir. Çapraz-modal özellik hizalaması gibi teknikler sayesinde mRNABERT, mRNA dizilerinin işlevselliğini yöneten karmaşık kurallara hakim olmamıza bir adım daha yaklaşmamıza yardımcı olur.
Yöntemler
Eğitim veri kümeleri
Veritabanı oluşturma ve veri toplama
RNA molekülleri iki kategoriye ayrılır: haberci RNA (mRNA) ve kodlamayan RNA (ncRNA). Ocak 2023 itibarıyla RNAcentral, 56 uzman veritabanından veri entegre eden ve 30 milyondan fazla dizi içeren en kapsamlı ve kapsamlı ncRNA veritabanıdır 38 . Mevcut RNA modelleri ağırlıklı olarak RNAcentral verileri üzerinde önceden eğitilmiştir. Ancak, büyük dil modellerini eğitmek için özel olarak tasarlanmış özel bir mRNA veritabanı mevcuttur. Bu boşluğu gidermek için, son çalışmalara benzer şekilde kapsamlı bir veri toplama süreci yürüterek bir mRNA veritabanı oluşturmaya başladık 85 .
Veri setimizin entegrasyonu için, NCBI 86’dan nt veritabanı ve MG-RAST 87 , GWH 88 , 89 ve MGnify 90’dan transkriptom birleştirme verileri de dahil olmak üzere çeşitli kaynaklardan mRNA verilerini topladık . Bu çeşitli kaynaklardan, yaklaşık 36 milyon diziden oluşan bir veri seti derlemek için tüm tam olgun mRNA dizilerini (tam CDS bölgeleri içeren) çıkardık.
Verilerin ön işlenmesi
Tutarlılığı sağlamak ve sonraki analizleri kolaylaştırmak için toplanan tüm dizileri DNA alfabesine eşledik. NCBI verileri (örneğin RefSeq veya GenBank) veri setimizin %70’inden fazlasını oluşturur ve birincil, yüksek kaliteli ve elle ek açıklamalı veri kaynağımız olarak hizmet eder. Sağlanan CDS konum ek açıklamalarını doğrudan kullandık. Kesin başlangıç konumlarının belirlenemediği kalan ek açıklama yapılmamış veriler için en doğru ve verimli açık okuma çerçevesi (ORF) tahmin yöntemini belirlemeye çalıştık. Temsili bir veri setinde yaygın olarak kullanılan birkaç aracı karşılaştırdık ve NCBI’ın ORFfinder 91’inin %40 CDS uzunluk filtresiyle birleştirildiğinde çeşitli türler arasında yüksek doğruluk ve üstün hesaplama verimliliği arasında en iyi dengeyi gösterdiğini gördük (Ek Tablo 1 ). Bu nedenle, her bir ORF’yi başlangıç kodonu ATG’den en yakın durdurma kodonuna kadar tanımlayarak bu yöntemi benimsedik ve en uzun sürekli bölge kodlama dizisi olarak belirlendi. Daha sonra, uzunluk ve yedeklilik kontrol ölçümleri gerçekleştirdik. Tahmini CDS uzunlukları mümkün olan maksimum değerin %40’ının altında olan diziler hariç tutulmuş ve yüksek kimlik fazlalığı gösterenler çıkarılmıştır. Ayrıca, hesaplama verimliliği ile veri kalitesi arasında denge sağlamak için, teorik maksimum uzunluk 3066 nükleotit olmasına rağmen, kodlama sonrası 1022 nükleotidi aşan diziler hariç tutulmuştur. Daha fazla ayrıntı ve tartışma, Ek Bilgiler Bölüm 2’de bulunabilir . Eğitim Öncesi Veri Setinin Analizi.
Daha sonra, yaklaşık 18 milyon benzersiz mRNA dizisinden oluşan yüksek kaliteli bir mRNA veri kümesini titizlikle oluşturduk. Her dizi, köken türüne göre sistematik olarak sınıflandırıldı ve bu da veri kökeninin daha ayrıntılı bir şekilde anlaşılmasını sağladı. Ayrıntılı bilgiler Ek Şekil 1 ve Ek Tablolar 2 ve 3’te sunulmaktadır . Model performansını ve genelleştirilebilirliğini titizlikle değerlendirmek için verileri rastgele katmanlara ayırdık ve bağımsız bir veri kümesi oluşturduk.
Model detayları
Dizi belirteçleme
Önceki mRNA ön eğitim modelleri, verilere ve hedef bölgeye bağlı olarak genellikle iki farklı kodlama yöntemi kullanıyordu: UTR’ler için tasarlanan modeller her bir nükleotidi bir belirteç olarak kodluyordu 40 , CDS modelleri ise belirteç olarak nükleotid üçlülerini (kodonları) kullanıyordu 42 , 43. Bu çalışmada, farklı belirteçleme yöntemlerini UTR’lere ve CDS’lere uygulayarak bu yaklaşımları birleştirdik. Bu yenilikçi yöntem, her bir parçanın ve tam dizinin gizli durumlarını ve dikkat ağırlıklarını yakalamamızı sağladı. Dizi alfabesini standartlaştırmak için, RNA-cDNA dizileme protokollerine uyması için tüm urasil (U) bazlarını timine (T) dönüştürdük. Giriş dizisi, her belirteç bir nükleotid, kodon veya özel karakteri temsil eden bir tam sayı olan T belirteçlerinden oluşan bir vektördür. Kelime dağarcığı 64 kodon, 5 nükleotid (A, T, C, G ve nadir baz N) ve beş özel belirteç içerir: maskeleme için [MASK], dolgu için [PAD], bilinmeyen kodonlar için [UNK] ve dizi sınırlarını belirtmek için [CLS] ve [SEP]. Modelin herhangi bir etiket veya ön bilgiyle eğitilmemiş olması, 6 numaralı belirtecin nükleotid A’yı mı yoksa 18 numaralı belirtecin başlangıç kodonu ATG’yi mi temsil ettiğini ayırt edemeyeceğini vurgulamaktadır.
Model mimarisi
mRNABERT, 768 gizli durum boyutuna sahip 12 dönüştürücü katmandan oluşur. DNABERT-230 ile benzer şekilde yapılandırılan mRNABERT , konumsal yerleştirmeleri Doğrusal Önyargılı Dikkat (ALiBi) ile değiştirir ve standart dikkat hesaplamalarının doğruluğunu artırırken zaman ve bellek açısından verimliliği artırmak için G/Ç farkında Flaş dikkati entegre eder. Jeton yerleştirme işleminden sonra, mRNA dizileri Dönüştürücüye girilir.(1)
Burada L, belirteç dizisinin uzunluğunu temsil eder.
Model eğitimi
Modeli, %15 maskeleme oranıyla MLM kaybını kullanarak önceden eğittik. Belirtmek gerekirse, belirteçlerin %80’i [MASK] belirteciyle değiştirildi, %10’u rastgele başka bir belirteçle değiştirildi ve kalan %10’u değiştirilmeden bırakıldı. Eğitim süresi ve maliyeti arasında denge kurmak için, dizi uzunluğunu maksimum 1022 belirteçle sınırladık; bu sayının çoğu mRNA transkriptini kapsayacak kadar yeterli olduğunu gördük. Her dizi grubu maksimum uzunluğa kadar dolduruldu.
Optimizasyon amacıyla, diğer parametreler için varsayılan ayarlarla birlikte 1 × 10 -4 öğrenme oranıyla AdamW optimize edicisini uyguladık . Öğrenme oranı, ilk 10.000 adımda 0’dan 1 × 10 -4’e doğrusal bir artış gösterdi ve ardından 1.000.000 adımda doğrusal bir düşüşle sıfıra indi. Eğitim ilerlemesini izlemek için, eğitim setinin %1’ini rastgele bir doğrulama seti olarak ayırdık. Bildirilen model, 10 dönem boyunca 660.000 gradyan adımına karşılık gelen NVIDIA A6000 GPU’larda eğitildi. 10.000 adımda doğrulama kaybında herhangi bir iyileşme gözlemledikten sonra eğitimi manuel olarak durdurduk. Eğitim kaybı eğrisi, Ek Şekil 3’te referans olarak mevcuttur .
Daha kapsamlı dizi bilgisi eklemek ve modelin çok modlu moleküler etkileşimleri öğrenmesini sağlamak için, MLM’den sonra karşılaştırmalı öğrenme gerçekleştirdik. Eğitim setinden 500.000 CDS verisi seçtik ve çevrilmiş amino asit dizilerini bir protein dili modeline (pLM) girdik. Performanstan ödün vermeden hesaplama verimliliğini optimize etmek için, amino asit veya protein gömmeleri oluşturmak üzere ProtT5-XL-UniRef50 model 26’nın yarı hassasiyetli sürümünü kullandık ve böylece GPU bellek tüketimini azalttık. pLM ağırlıkları dondurulmuş halde tutularak, dizileri ilgili modellere besledik ve gömmeleri son gizli katmandan çıkardık. Dizileri sabit boyutlu vektörler (CDS için 768 boyut ve amino asitler için 1024 boyut) olarak kodlamak için elde edilen gömmelerin ortalamasını aldık. Karşılaştırmalı öğrenmeyi kolaylaştırmak için, 768 ve 1024 boyutlu vektörleri 256 boyuta yansıtmak için eğitilebilir bir doğrusal katman kullanıldı. OpenAI-CLIP kütüphanesi 92’nin kullanımıyla , modeli, karşılık gelen diziler arasındaki mesafeyi en aza indirirken, karşılık gelmeyen diziler arasındaki mesafeyi en üst düzeye çıkarmayı amaçlayan karşılaştırmalı bir kayıp fonksiyonuyla eğittik. Bu amaçla kullanılan kayıp fonksiyonu aşağıdaki gibi tanımlanmıştır : 93(2)(3)(4)(5)(6)(7)(8)(9)(10)
Burada C, CDS dizi yerleştirmelerini, A ise amino asit dizi yerleştirmelerini temsil eder ve her ikisi de 256 boyuta yansıtılmıştır. Son eğitilmiş model mRNABERT’tir.
Model değerlendirmesi
Görselleştirme için belirteç ve dizi yerleştirmelerinin boyutunu iki boyuta indirmek amacıyla t-SNE yöntemi 60’ı kullandık . Başlangıçta, tüm kelime dağarcığı sözcüklerine ait yerleştirmeler çıkarıldı ve amino asit kategorilerine ve özelliklerine göre kümelendi. ARI ve FMI’yi hesaplamak için sırasıyla Scikit-learn kütüphanesindeki adjusted_rand_score ve fowlkes_mallows_score fonksiyonlarını kullandık. Bu metrikler şunlara dayanmaktadır:(11)(12)(13)
Daha sonra, alt akış veri kümelerinden 5’UTR, CDS ve 3’UTR’den rastgele segmentler seçtik ve doğrulama kümesinden tam mRNA dizileri seçildi. Uzun kodlamayan RNA (lncRNA) verileri GENCODE veritabanından elde edildi 94. RNA sınıflandırması için toplam 9287 giriş kullanıldı. Son olarak, tür sınıflandırması için toplam 3452 tam mRNA dizisi girişi kullanarak memelileri, böcekleri, bitkileri, bakterileri, mantarları ve virüsleri kapsayan doğrulama veri kümesinden altı temsili tür seçtik. Tüm veriler, eğitim kümesinden kimlik ve dışlanmada %40’tan az örtüşme sağlamak için dikkatlice düzenlendi.
Aşağı akış görev veri kümeleri
5′ UTR dizilerinin ribozom yükleme veri kümeleri
Veri seti, Sample ve ark. tarafından yürütülen bir MPRA araştırmasından türetilmiştir. 50 , ilgili ortalama ribozom yükleriyle eşleştirilmiş rastgele 50 nükleotid uzunluğunda 5′ UTR dizilerinden oluşmaktadır. Veri güvenilirliğini artırmak için okuma sayıları eklenmiştir. Veri seti sekiz kütüphaneye ayrılmıştır ve bunlar da iki gruba ayrılmıştır. Altı kütüphane, geliştirilmiş yeşil floresan proteinini (eGFP) kodlayan sabit bir bölge içerirken, kalan iki kütüphane eGFP yerine mCherry’nin kodlama dizilerine (CDS) sahiptir. eGFP grubu içinde, değiştirilmemiş üridin (U) içeren iki kütüphane ve psödouridin (Ψ) ve 1-metilpsödouridin (m1Ψ) ile değiştirilmiş kütüphaneler bulunmaktadır. Veri seti bölme ve ince ayar stratejilerinin ayrıntıları ek bilgilerde bulunabilir.
CDS dizileriyle ilgili veri kümeleri
Eş anlamlı kodon randomizasyonu yoluyla oluşturulan mRFP veri kümesi 61 , kırmızı floresan proteininin (mRFP) tam kodlama dizisini kapsar ve E. coli’deki 1459 gen varyantından oluşur . Protein verimini kaydeder ve kodon kullanımı ile protein üretimi arasındaki ilişkiyi araştırır. Mantar veri kümesi 62 , birden fazla türe ait çeşitli mantar genomlarından protein kodlayan genler ve tRNA genleri hakkında bilgi derler. E. coli veri kümesi 63 , sırasıyla 2308, 2067 ve 1973 dizileriyle düşük, orta ve yüksek ifade seviyelerine kategorize edilmiş, E. coli’deki protein ekspresyonu hakkında deneysel veriler içerir . mRNA stabilitesi veri kümesi 64 , zebra balığı, Xenopus laevis embriyoları ve fare ve insan hücrelerinden mRNA stabilitesi özelliklerine ilişkin bilgiler sağlayarak, mRNA stabilitesinin kodona bağlı düzenlenmesini açıklamayı amaçlamaktadır. Tc-Riboswitches veri seti 66, GFP’nin yukarısında konumlandırılmış tetrasiklin (Tc) riboswitch dimer dizilerini içermekte olup, Tc’nin varlığında veya yokluğunda farklı etkileri ayırt etmek için anahtar faktörünün değerlendirilmesini kolaylaştırmaktadır. SARS-CoV-2 Aşı Degradasyonu veri seti 65, yapısal özellikler, kararlılık ve translasyon verimliliğinden türetilen optimize edilmiş mRNA dizilerini içermektedir. Tutarlı bir veri bölümleme stratejisi kullanarak, mRNABERT modelini geliştirdik ve mevcut literatürle karşılaştırmalı bir analiz gerçekleştirdik. Bu veri setleri hakkında daha fazla ayrıntı ek bilgilerde sunulmaktadır.
Dokuz hücre hattında RBP bağlanma bölgeleri ve insan m6A modifikasyonları
Yang ve ark.’dan iki veri seti topladık ve analiz ettik. 41 İlk veri seti, 19 RBP’yi kapsayan 31 CLIP (çapraz bağlama immünopresipitasyon) deneyinden elde edilen verileri bir araya getiriyor. Bu verileri analiz etmek için, pozitif veri setleri (RBP bağlanma bölgeleri) ile negatif veri setleri (bağlanmayan bölgeler) arasında ayrım yapan birleşik bir veri işleme iş akışı ve belirli dizi pencere boyutları kullanıldı. Yan yana bölgeler arasında gereksiz tekrarları azaltmak ve etkileşimi önlemek için önlemler alındı.
İkinci veri seti, dokuz hücre hattında tek nükleotid çözünürlüğünde insan m 6 A modifikasyon verilerini içermekte olup, 131.703 yüksek güvenilirlikli m 6 A bölgesi ile sonuçlanmıştır. Aynı transkriptlerin 3′ UTR’sinden m 6 A olmayan adenozinleri seçip kopyaları çıkararak, 79.021 m 6 A bölgesi ve 849.005 m 6 A olmayan bölge içeren bir veri seti oluşturduk ve 1:10 pozitif/negatif oranını koruduk. Doğruluk ve güvenilirliği sağlamak için 10 katlı çapraz doğrulama ve rastgele aşağı örnekleme yöntemleri kullandık.
Ekleme yerleri ve alternatif poliadenilasyon veri kümeleri
Kullanılan ekleme yeri veri kümesi GS_1 veri kümesi 95’ti . Bu veri kümesi, negatif örneklerin ekzon, intron veya yanlış pozitif dizilerden oluştuğu, pozitif ve negatif örneklerin dengeli bir oranını korur. Veri kümesi, G3PO+ genomik dizilerinin ekzon ve intron bölgelerinden rastgele diziler seçilerek oluşturuldu. İnsanlar da dahil olmak üzere 148 ökaryotik organizmadan türetilen hatasız ekleme yeri dizilerini içerir. Daha da önemlisi, test veri kümesi, eğitim setinde temsil edilmeyen dört farklı türden dizileri içermektedir. Kullanılan APA veri kümesi, Bogard’ın 96 veri kümesindeki 3 milyondan fazla APA raporlayıcı gen girişinden 228.000 diziyi filtreleyen BEACON’dan kaynaklanmıştır. Bu regresyon görevi, proksimal APA izoformlarının göreceli oranını ölçmeyi ve değerlendirmeyi amaçlamaktadır.
Protein mühendisliği görev veri kümeleri
Gömme işleminin kalitesini doğrulamak için Carlos ve arkadaşları tarafından oluşturulan birkaç protein mühendisliği görev veri kümesi kullanıldı . 42 Bu veri kümeleri, FLIP 97 çalışmasında bildirilen bir dizi erime sıcaklığını ve Sridharan ve arkadaşları tarafından yürütülen çözünürlük deneylerinden elde edilen çözünürlük vekil verilerini kapsamaktadır. 98 Amino asit dizileri, UniProt kimlikleri kullanılarak nükleotid dizilerine eşlendi; eşlenemeyen veya standartları karşılamayan diziler hariç tutuldu. Ayrıca, yedi model organizmadan alınan RNA dizilerinden oluşan transkriptom verileri, milyon başına transkriptler aracılığıyla derlemelerdeki transkript bolluğunu tahmin etmek ve bu verileri mevcut dizi veritabanlarına eşlemek için kullanıldı.
Tam uzunlukta mRNA dizilerinin veri kümesi
Leppek ve arkadaşları tarafından yürütülen PERSIST-seq çalışması23, çeşitli mRNA dizilerinin hücre içi ve hücre dışı translasyon verimliliğini ve kararlılığını sistematik olarak değerlendiren tam uzunlukta mRNA veri setini sağlar. mRNA ‘kütüphanesi, 112 benzersiz 5′ ve/veya 3’ UTR’ye sahip 233 farklı mRNA dizisi içerir. Sample ve arkadaşlarının çalışmasında taranan randomize kısa UTR’lerin aksine50 , bu kütüphane hücresel ve viral genomlardan gelen diziler dahil olmak üzere mRNA ekspresyonunu test etmek için tam uzunlukta doğal UTR’leri kullanır. Çeşitli algoritmalar ve tasarım yöntemleri kullanılarak22 , 99 , CDS dizileri ve protein hedeflerinin yapıları çeşitlendirildi ve 121 CDS varyantı elde edildi. PERSIST-seq, bu mRNA kütüphanesindeki yapıların polisom profillerini analiz ederek ribozom yükünü ve kararlılığını hesapladı ve farklı işlevsel bölgelerin mRNA özellikleri üzerindeki etkilerinin kapsamlı bir şekilde anlaşılmasına yardımcı oldu. Bu veri kümeleri, mRNABERT modelimizi hassas bir şekilde ayarlamak için kullanıldı. Kıyaslama modelleriyle adil bir performans karşılaştırması sağlamak için, başlangıçta raporlarında hassas ayar yöntemleri kullanmayan bazı modellere rağmen, Hugging Face’in 100 numaralı çok moleküllü kütüphanesini kullanarak diğer modellerde de hassas ayar yaptık .
Polisom ve ribozom profillemesi, çeviri hızlarını değerlendirmek için doğrudan yöntemler olarak belirlenmiştir 101. Zheng ve ark. 84, 3819 ribozomal profilleme veri setinden oluşan kapsamlı bir koleksiyonu derleyip değerlendirerek, bunları 140’tan fazla insan ve fare hücre tipini kapsayan transkriptom çapında bir çeviri verimliliği (TE) ölçümleri atlasına dönüştürdüler. Bu kaynağı, dizi kodlu mRNA özelliklerine dayanarak yüzlerce hücre tipindeki TE’leri tahmin etmek için kullandık.
Her bir alt akış görevi için kıyaslama modelleri ve metodolojilerinin ayrıntılı açıklamaları ek bilgilerde sunulmaktadır.
Raporlama özeti
Araştırma tasarımına ilişkin daha fazla bilgi , bu makaleye bağlantısı verilen Nature Portfolio Raporlama Özetinde mevcuttur.